
Tłumaczył dr n. med. Dariusz Stencel
Konsultowała prof. dr hab. n. med. Przemysława Jarosz-Chobot, Klinika Diabetologii Dziecięcej Śląskiego Uniwersytetu Medycznego w Katowicach
Skróty: BOHB – kwas ß-hydroksymasłowy, DKA – cukrzycowa kwasica ketonowa, HHS – stan hiperglikemiczno-hiperosmolalny
Podsumowanie i zalecenia
Kryteria biochemiczne rozpoznania cukrzycowej kwasicy ketonowej (diabetic ketoacidosis – DKA) są następujące:
– hiperglikemia (stężenie glukozy we krwi >200 mg/dl [11 mmol/l])
– pH krwi żylnej <7,3 lub stężenie wodorowęglanu <15 mmol/l
– ketonemia i ketonuria.
Objawy kliniczne DKA obejmują:
– odwodnienie (bywa trudne do rozpoznania)
– tachykardię
– zwiększenie częstotliwości oddechów – tachypnöe (może sugerować zapalenie płuc lub astmę)
– przyśpieszony, głęboki oddech (oddech Kussmaula); wydychane powietrze ma zapach acetonu (określany też jako zapach zmywacza do paznokci lub fermentujących owoców)
– nudności, wymioty (mogą sugerować nieżyt żołądkowo-jelitowy)
– ból brzucha przypominający objawy ostrego brzucha
– splątanie, senność, postępujące ograniczenie świadomości i w końcu całkowitą utratę przytomności.
Czynniki ryzyka DKA u dzieci z nowo rozpoznaną cukrzycą obejmują: młodszy wiek (<2 lat), opóźnienie rozpoznania, niższy status społeczno-ekonomiczny oraz zamieszkanie w kraju charakteryzującym się rzadkim występowaniem cukrzycy typu 1.
Czynniki ryzyka DKA u chorych z wcześniej rozpoznaną cukrzycą obejmują: pominięcie dawki insuliny, niedostateczną kontrolę metaboliczną, epizody DKA w przeszłości, nieżyt żołądkowo-jelitowy z utrzymującymi się wymiotami i trudnością z zapewnieniem odpowiedniego poziomu nawodnienia, zaburzenia psychiatryczne (w tym zaburzenia odżywiania), trudne warunki społeczne i rodzinne, wiek okołopokwitaniowy i młodzieńczy u dziewcząt, utrudniony dostęp do opieki medycznej, błędy związane ze stosowaniem osobistej pompy insulinowej.
Przedstawione zalecenia, oparte na aktualnie dostępnych wiarygodnych danych, są jedynie ogólnymi wskazówkami dotyczącymi leczenia DKA. Z uwagi na znaczne różnice między objawami DKA u poszczególnych chorych (od łagodnych objawów z niewielkim odwodnieniem do ciężkiego przebiegu ze znacznym odwodnieniem), niektórzy mogą wymagać szczególnych metod leczenia. Lekarz ma za zadanie ocenić, czy w konkretnym przypadku przedstawione tutaj opcje są wystarczające, czy też chory wymaga postępowania wykraczającego poza zakres wytycznych. Wybór optymalnej tera- pii u poszczególnych chorych powinien się opierać na ocenie klinicznej, a wszystkie zmiany w trakcie leczenia (dawki insuliny, skład elektrolitów oraz objętość płynów nawadniających) powinny być efektem ścisłego monitorowania odpowiedzi klinicznej i biochemicznej.
- Ocenę w stanie zagrożenia należy przeprowadzić zgodnie z wytycznymi Pediatric Advanced Life Support (PALS), uwzględniając: natychmiastowe oznaczenie stężenia glukozy we krwi, oznaczenie stężenia związków ketonowych we krwi lub moczu, stężenia elektrolitów w surowicy, gazometrii oraz morfologii krwi obwodowej, ocenę stopnia odwodnienia oraz ocenę poziomu świadomości (E). Należy wprowadzić drugą kaniulę do żył obwodowych (E).
- Leczenie należy prowadzić w ośrodkach z doświadczeniem w terapii DKA u dzieci i młodzieży, w których możliwe jest częste monitorowanie podstawowych parametrów życiowych, stanu neurologicznego oraz wyników badań laboratoryjnych (E). Jeśli ze względu na ograniczenia geograficzne leczenie rozpoczyna się w ośrodku o mniejszym doświadczeniu, o ograniczonych możliwościach terapeutycznych, należy zapewnić kontakt telefoniczny lub organizację telekonferencji z lekarzem doświadczonym w leczeniu DKA (E).
- Leczenie można odpowiednio szybko zmodyfikować tylko na podstawie objawów klinicznych i wyników badań laboratoryjnych, dlatego konieczne jest ścisłe monitorowanie stanu klinicznego i biochemicznego pacjenta (E).
-
Celem leczenia jest przywrócenie odpowiedniego nawodnienia, wyrównanie kwasicy i odwrócenie ke- tozy, stopniowe wyrównanie stanu hiperosmolalności oraz przywrócenie prawidłowego stężenia glukozy we krwi, monitorowanie i leczenie powikłań DKA oraz rozpoznanie i leczenie stanów wywołujących DKA.
-
Uzupełnianie płynów należy rozpocząć przed pierwszym podaniem insuliny. Objętość podawanych płynów należy zwiększać w zależności od potrzeby, aż do przywrócenia prawidłowego krążenia obwodowego (E). Tempo dalszego podawania płynów należy obliczyć w taki sposób, aby uwzględnić pod- stawowe zapotrzebowanie płynowe oraz wyrównać niedobór wody równomiernie w ciągu 48 h. Objętość podanych płynów rzadko przekracza 1,5–2-krotnie zwykłe dobowe zapotrzebowanie (C).
- Leczenie insuliną. Należy rozpocząć wlew insuliny w dawce 0,05–0,1 j./kg mc./h 1–2 h po rozpoczęciu uzupełniania płynów (C, B).
- Potas. W przypadku hiperkaliemii należy opóźnić suplementację potasu do momentu udokumentowania diurezy. W pozostałych przypadkach początkowe stężenie potasu we wlewie kroplowym powinno wynosić 40 mmol/l lub 20 mmol/l, jeżeli niedobór płynów jest uzupełniany w tempie >10 ml/kg mc./h (E).
- Nie zaleca się podawania wodorowęglanu, z wyjątkiem leczenia zagrażającej życiu hiperkaliemii (B).
- Przedmiotowe i podmiotowe objawy ostrzegawcze obrzęku mózgu obejmują: ból głowy (o zróżnicowanym nasileniu) i zmniejszenie częstotliwości rytmu serca, zmiany stanu neurologicznego (niepokój, drażliwość, nasilona senność, nietrzymanie moczu i/lub stolca), swoiste objawy neurologiczne (np. porażenie nerwów czaszkowych), zwiększenie ciśnienia tętniczego i zmniejszenie wysycenia hemoglobiny tlenem.
- Jeżeli stwierdza się wiele czynników ryzyka obrzęku mózgu, przy łóżku chorego powinien być dostępny mannitol lub hipertoniczny roztwór NaCl do ewentualnego podania dożylnie we wcześniej obliczonej dawce (E). Jeśli stan neurologiczny chorego znacznie się pogarsza, należy natychmiast rozpocząć podawanie płynów hiperosmolalnych (C).
- Zapobieganie. Postępowanie w przypadku DKA można uznać za kompleksowe dopiero po zidentyfikowaniu przyczyny tego epizodu i podjęciu próby leczenia przyczynowego. Nawracające epizody DKA bez poprzedzającej gorączki lub wymiotów są prawie wyłącznie wynikiem problemów psychospołecznych oraz pominięcia dawki insuliny (E).
- Kryteria rozpoznania stanu hiperglikemiczno-hiperosmolalnego (hyperglycemic hyperosmolar state – HHS) obejmują:
– stężenie glukozy w osoczu >600 mg/dl (33,3 mmol/l)
– pH krwi żylnej >7,25; pH krwi tętniczej >7,30
– stężenie wodorowęglanu w surowicy >15 mmol/l
– niewielką ketonurię, nieobecną lub łagodną ketonemię
- efektywną osmolalność osocza >320 mOsm/kg
– zmianę stanu świadomości (np. ograniczona przytomność, agresja) lub drgawki.
- Celem początkowej terapii płynowej w HHS jest zwiększenie objętości płynu wewnątrz- i zewnątrznaczyniowego, przywrócenie prawidłowego przepływu nerkowego oraz stopniowe zmniejszenie stężenia sodu w surowicy i osmolalności.
- Podawanie insuliny w HHS należy rozpocząć od dawki między 0,025 a 0,05 j./kg mc./h, jeśli w trakcie stosowania samej terapii płynowej stężenie glukozy we krwi zmniejsza się wolniej niż co najmniej o 50 mg/dl (3 mmol/l/h [C]).
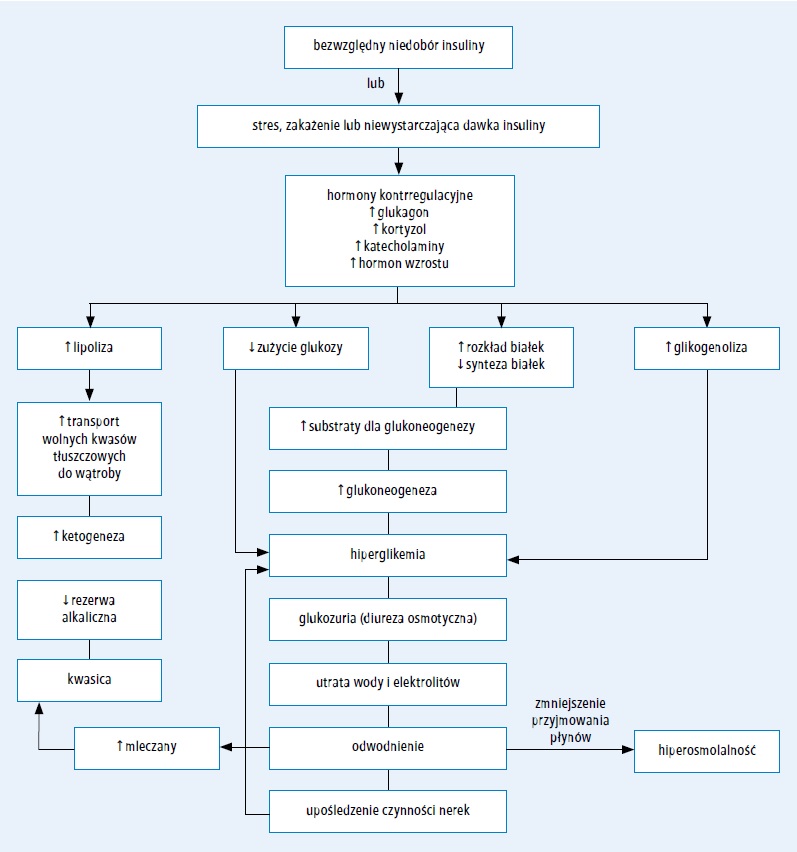
Ryc. 1. Patofizjologia cukrzycowej kwasicy ketonowej (przedrukowano za zgodą z 232. pozycji piśmiennictwa)
Przyczyną DKA jest niedobór krążącej insuliny oraz zwiększone stężenie hormonów kontrregulacyjnych: katecholamin, glukagonu, kortyzolu i hormonu wzrostu.1,2 Ciężki niedobór insuliny występuje we wcześniej nierozpoznanej cukrzycy typu 1 oraz u pacjentów, którzy celowo lub nieumyślnie nie przyjmują insuliny, szczególnie insuliny długo działającej w schemacie intensywnej insulinoterapii. U chorych stosujących osobiste pompy insulinowe może dojść do szybkiego rozwoju DKA w przypadku zatrzymania wlewu insuliny z jakiegokolwiek powodu.3 Przyczyną względnego niedoboru insuliny jest zwiększenie stężenia hormonów antagonistycznych w odpowiedzi na stres w takich stanach, jak posocznica, uraz lub choroby układu pokarmowego z towarzyszącymi wymiotami i biegunką, co uniemożliwia utrzymanie homeostazy i prowadzi do zaburzenia równowagi metabolicznej pomimo przyjęcia zalecanej dawki insuliny.
Względny lub bezwzględny niedobór insuliny w skojarzeniu z dużymi stężeniami hormonów kontrregulacyjnych prowadzi do przyspieszonego katabolizmu ze zwiększeniem produkcji glukozy w wątrobie i nerkach (poprzez glikogenolizę i glukoneogenezę) oraz upośledzenia obwodowego zużycia glukozy, czego skutkiem jest hiperglikemia i hiperosmolalność. Niedobór insuliny i duże stężenia hormonów kontrregulacyjnych nasilają także lipolizę i ketogenezę, co powoduje ketonemię i kwa- sicę metaboliczną. Hiperglikemia przekraczająca próg nerkowy wynoszący zwykle około 180 mg/dl (10 mmol/l; zakres progu nerkowego u chorych na cukrzycę i osób zdrowych jest bardzo szeroki) i hiperketonemia prowadzą do diurezy osmotycznej, odwodnienia i nieuniknionej utraty elektrolitów, często nasilanej przez wymioty wywołane ciężką ketonemią. Zaburzenia te stymulują dalszą produkcję hormonów stresowych, co jeszcze bardziej nasila insulinooporność oraz hiperglikemię i hiperketonemię. W celu przerwania tego błędnego koła należy podać egzogenną insulinę, płyny i elektrolity, w przeciwnym razie może dojść do zgonu z powodu odwodnienia i kwasicy metabolicznej. DKA może nasilać kwasica mleczanowa wynikająca z zaburzenia perfuzji tkanek i/lub posocznicy.
DKA charakteryzuje się znaczną utratą wody i elektrolitów z przestrzeni wewnątrz - i zewnątrzkomórkowej (p. tab. 1. i ramka 1.). Mimo odwodnienia ciśnienie tętnicze utrzymuje się jednak na prawidłowym lub nawet wyższym poziomie,5 prawdopodobnie w związku ze zwiększonym stężeniem katecholamin, zwiększonym uwalnianiem hormonu antydiuretycznego (ADH) w odpowiedzi na hiperosmolalność, prowadzącym do zwiększenia ciśnienia krwi poprzez receptory V2 lub w wyniku działania innych czynników.5 W związku z glukozurią chorzy oddają mocz w dużej objętości, do czasu wystąpienia krańcowo dużego niedoboru płynów oraz wstrząsu prowadzącego do krytycznego zmniejszenia przepływu nerkowego i filtracji kłębuszkowej. Nasilenie poszczególnych nie- doborów w chwili pierwszej oceny chorego zależy od czasu trwania i ciężkości choroby, zdolności pacjenta do przyjmowania płynów i elektrolitów oraz składu posiłków i płynów spożytych przed zgłoszeniem się do lekarza. Przyjmowanie płynów bogatych w węglowodany (soki lub napoje zawierające cukier) nasila hiperglikemię.6 Gwałtowne monu opróżnienie żołądka z treści zawierającej dużą ilość cukru, co wynika z ustąpienia gastroparezy w przebiegu leczenia, u niektórych pacjentów powoduje zwiększenie stężenia glukozy we krwi zaraz po rozpoczęciu leczenia mimo jej znacznej utraty z moczem.7
W tabeli 2. wymieniono objętość płynów konieczną do zaspokojenia zapotrzebowania podstawowego i wyrównania niedoboru wody u chorych z DKA według Darrowa (w zależności od masy ciała, przy założeniu 10% odwodnienia).16
Ramka 1. Objawy kliniczne DKA
• odwodnienie
• tachypnöe oraz przyśpieszony, głęboki oddech (oddech Kussmaula)
• nudności, wymioty i ból brzucha przypominający „ostry brzuch”
• splątanie, senność, postępujące zaburzenia świadomości i utrata przytomności
Definicja DKA
• Kryteria biochemiczne:17
– hiperglikemia (stężenie glukozy we krwi >200 mg/dl [11 mmol/l])
– pH krwi żylnej <7,3 lub stężenie wodorowęglanu <15 mmol/l
– ketonemiaa i ketonuria.
Związki ketonowe są zwykle obecne w moczu w stężeniu ≥2+ („umiarkowana” lub „ciężka” ketonuria). U dzieci, u których rozpoczęto już leczenie, oraz u tych, które spożywały niewielką ilość węglowodanów lub nie spożywały ich wcale, czasami obserwuje się jedynie umiarkowane zwiększenie stężenia glukozy we krwi określone jako „DKA z euglikemią”.19,20
U dzieci coraz częściej występuje cukrzyca typu 2. Na całym świecie zapadalność i występowanie cukrzycy typu 2 u dzieci i młodzieży znacznie się różni, w zależności od kraju, przedziału wiekowego oraz pochodzenia etnicznego, co można tłumaczyć różnicami dotyczącymi charakterystyki badanej populacji oraz odmiennościami metodyki poszczególnych badaniach.21 W niektórych amerykańskich ośrodkach cukrzyca typu 2 stanowi obecnie aż połowę przypadków nowo rozpoznawa- nej cukrzycy u osób w wieku 10–21 lat.22 W amerykańskim badaniu Diabetes in Youth Study wykazano, że w momencie rozpoznania cukrzycy typu 2 DKA stwierdzono u prawie 10% chorych,23 jednak ogółem DKA występuje u 5–25% chorych jako pierwsza manifestacja cukrzycy typu 2.24
• Stopień ciężkości DKA określa się w zależności od stopnia nasilenia kwasicy25:
– lekka: pH krwi żylnej <7,3 lub stężenie wodorowęglanu <15 mmol/l
– umiarkowana: pH <7,2, stężenie wodorowęglanu <10 mmol/l
– ciężka: pH <7,1, stężenie wodorowęglanu <5 mmol/l.
• HHS, określany wcześniej jako nieketonowa śpiączka hiperosmolalna, może wystąpić u młodych chorych na cukrzycę typu 2,26-28 chorych na cukrzycę typu 129 oraz u niemowląt, zwłaszcza w przebiegu przemijającej cukrzycy noworodków związanej z mutacją 6q24.30 Kryteria rozpoznania HHS obejmują:31,32
– stężenie glukozy w osoczu >600 mg/dl (33,3 mmol/l)
– pH krwi tętniczej >7,30, pH krwi żylnej >7,25
– stężenie wodorowęglanu w osoczu >15 mmol/l
– niewielka ketonuria, brak ketonemii lub niewielka ketonemia (oznaczane metodą reakcji z nitroprusydkiem)
– efektywna osmolalność osocza >320 mOsm/kg
– ograniczenie świadomości, agresja lub drgawki (u ok. 50% chorych).
Należy pamiętać, że charakterystyczne objawy HHS i DKA mogą się na siebie nakładać. U niektórych chorych z HHS, zwłaszcza z ciężkim odwodnieniem, można stwierdzić lekką lub umiarkowaną kwasicę, wynikającą głównie ze zmniejszonej perfuzji tkankowej i/lub kwasicy mleczanowej. Z drugiej strony, niektóre dzieci chore na cukrzycę typu 1 mogą wykazywać objawy HHS (ciężka hiperglikemia), zwłaszcza jeżeli przed ustaleniem rozpoznania otrzymywały napoje o dużej zawartoś ci węglowodanów w celu zaspokojenia pragnienia i wyrównania utraty płynów z moczem.6Leczenie należy odpowiednio zmodyfikować zgodnie z patofizjologią i swoistymi zaburzeniami biochemicznymi u poszczególnych pacjentów (p. niżej). Zasady leczenia HHS opisano na stronie 203.
Częstość występowania DKA
W chwili ujawnienie się cukrzycy
Częstość występowania DKA w chwili ujawnienia się cukrzycy charakteryzuje się dużą zmiennością geograficzną, z odwrotną zależnością do zapadalności na cukrzycę typu 1 w danym regionie. W Europie i Ameryce Północnej DKA występuje u około 15–70% chorych.23,33-38 DKA w momencie rozpoznania cukrzycy częściej stwierdza się u młodszych dzieci (w wieku <2 lat), w następstwie błędów diagnostycznych lub opóźnionego leczenia,39-41 a także u dzieci z mniejszości etnicznych oraz z rodzin mających ograniczony dostęp do opieki medycznej ze względów społecznych lub ekonomicznych.20,23,37,39,42,43
Dzieci z wcześniej rozpoznaną cukrzycą
U chorego z rozpoznaną wcześniej cukrzycą typu 1 ryzyko wystąpienia DKA wynosi 1–10% na rok.3,44-48 Ryzyko jest większe u:47
– dzieci pomijających dawki insuliny46
– dzieci z niedostateczną kontrolą metaboliczną lub epizodami DKA w przeszłości
– chorych z towarzyszącym nieżytem żołądkowo-jelitowym z utrzymującymi się wymiotami, którym nie można zapewnić odpowiedniego nawodnienia
– dzieci z zaburzeniami psychicznymi, w tym zaburzeniami odżywiania
– dzieci z rodzin w trudnej lub niestabilnej sytuacji (np. nadużycia władzy rodzicielskiej)
– dziewcząt w okresie okołopokwitaniowym i w okresie pokwitania
– dzieci z ograniczonym dostępem do opieki medycznej
– chorych leczonych za pomocą osobistej pompy insulinowej (ponieważ pompy wykorzystują jedynie insulinę krótko działającą lub szybko działające analogi insuliny, przerwa w podawaniu insuliny z jakiegokolwiek powodu szybko prowadzi do jej niedoboru).3,49
Prawie wszystkie przypadki nawracającej DKA wynikają z pominięcia dawki insuliny lub niestosowania się do zaleceń postępowania w przypadku choroby towarzyszącej o ostrym przebiegu albo nieprzestrzegania instrukcji obsługi osobistej pompy insulinowej.
Postępowanie w DKA (ryc. 2.)
Ocena w stanie zagrożenia
W stanie zagrożenia należy postępować zgodnie z ogólnymi wytycznymi PALS.50,51 Poniżej wymieniono aspekty wymagające szczególnej uwagi.
• Natychmiast zmierz stężenie glukozy we krwi oraz stężenie BOHB (lub związków ketonowych) we krwi za pomocą aparatu przyłóżkowego. Konieczna jest też ocena kliniczna pacjenta w kierunku ewentualnego zakażenia.
– Pomiar stężenia BOHB we krwi przyłóżkowo (jeśli jest dostępny) jest przydatny do potwierdzenia DKA (u dzieci ≥3 mmol/l)18 i monitorowania odpowiedzi na leczenie.52-58
• Zważ chorego. Jeśli planujesz rozpoczęcie terapii płynowej dostosowanej do powierzchni ciała pacjenta, zmierz wysokość lub długość jego ciała. Do obliczeń wykorzystaj aktualną masę ciała, a nie stwierdzoną na poprzedniej wizycie w poradni lub odnotowaną w dokumentacji szpitalnej.
• Oceń stopień odwodnienia.
– Ocena kliniczna odwodnienia jest nieprecyzyjna. Zgodność takich ocen przeprowadzonych przez różnych lekarzy jest jedynie niewielka lub umiarkowana.59-61 Stopień odwodnienia należy ocenić na podstawie określonego zespołu objawów w badaniu przedmiotowym. Trzy pojedyncze objawy wskazujące na co najmniej 5% odwodnienie u dzieci w wieku od 1 miesiąca do 5 lat to:
– przedłużony nawrót kapilarny (norma: ≤1,5–2 s)
– nieprawidłowa elastyczność skóry (skóra „stojąca” lub nieelastyczna)
– nieprawidłowe oddychanie (hiperwentylacja).62
– Wśród innych objawów przydatnych w ocenie stopnia odwodnienia wymienia się między innymi: suchość błony śluzowej jamy ustnej, zapadnięcie gałek ocznych, brak wydzielania łez, osłabienie tętna i chłodne kończyny.
Większe nasilenie objawów zwykle wiąże się z cięższym odwodnieniem.62
– Do objawów co najmniej 10% odwodnienia należy osłabienie lub brak tętna obwodowego, niedociśnienie tętnicze i oliguria.
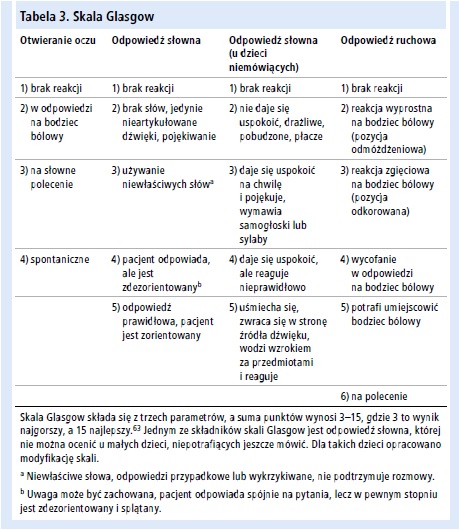
• Oceń poziom świadomości (skala Glasgow – tab. 3.).63
• Pobierz próbkę krwi w celu laboratoryjnego oznaczenia:
– stężenia glukozy w surowicy lub osoczu
– stężenia elektrolitów (w tym wodorowęglanu)
– stężenia mocznika i kreatyniny w surowicy krwi
– osmolalności surowicy – pH i pCO2 we krwi żylnej
– stężenia hemoglobiny, hematokrytu i morfologii krwi obwodowej (należy podkreślić, że zwiększona liczba leukocytów w odpowiedzi na stres jest charakterystyczną cechą DKA i nie zawsze wskazuje na towarzszące jej zakażenia)64
– stężenie albumin, wapnia, fosforu i magnezu.
• Oznaczenie HbA1c samo w sobie nie odgrywa istotnej roli w leczeniu DKA, może się jednak przydać w ocenie klinicznej i postępowaniu u niektórych chorych, ponieważ dostarcza informacji na temat czasu trwania hiperglikemii.
• Wykonać badanie moczu na obecność związków ketonowych.
• Odpowiednie próbki do badań mikrobiologicznych (krew, mocz, wymaz z gardła) pobierz jedynie w przypadku objawów wskazujących na zakażenie (np. gorączki).
• Jeżeli oznaczenie laboratoryjne stężenia potasu w surowicy opóźnia się, należy wykonać elektrokardiogram (EKG) w celu wyjściowej oceny kaliemii.65,66
Działania dodatkowe
Poniżej wymieniono aspekty, na które należy zwrócić szczególną uwagę u dzieci z przełomem hiperglikemicznym.
• U chorych nieprzytomnych lub ze znacznie ograniczoną świadomością zabezpiecz drożność dróg oddechowych i opróżnij żołądek za pomocą ciągłego odsysania przez zgłębnik wprowadzony przez nos, aby zapobiec aspiracji treści żołądkowej do płuc.
– W miarę możliwości należy unikać intubacji; nagłe zwiększenie ciśnienia parcjalnego CO2 w trakcie intubacji lub po jej wykonaniu może doprowadzić do zmniejszenia pH płynu mózgowo-rdzeniowego i nasilić obrzęk mózgu.67
• U chorych, którzy niedawno spożyli dużą objętość płynów zawierających glukozę, rozważ opróżnienie żołądka, nawet jeżeli nie stwierdzasz u nich zaburzeń świadomości.
– Po wypiciu dużej objętości soków owocowych lub słodzonych napojów żołądek może zawierać dużo wody z niewielką ilością sodu. Opróżnianie się żołądka we wczesnym okresie leczenia prowadzi do wchłaniania z przewodu pokarmowego glukozy i wody niezawierającej elektrolitów.7,68
• W przypadku ciężkiej niewydolności krążenia lub wstrząsu choremu należy podać do oddychania tlen.
• W celu oceny zmian załamków T w kierunku hiper- lub hipokaliemii choremu należy podłączyć kardiomonitor do ciągłego monitorowania EKG.65,66
• W celu zapewnienia możliwości wygodnego i bezbolesnego pobierania wielu próbek krwi, załóż choremu drugą obwodową kaniulę dożylną. Niektórzy pacjenci w bardzo ciężkim stanie, leczeni na oddziale intensywnej opieki medycznej, mogą wymagać założenia kaniuli dotętniczej.
– Poza absolutnie koniecznymi przypadkami należy unikać zakładania cewnika do żył głównych z uwagi na duże ryzyko zakrzepicy, szczególnie u bardzo małych dzieci. Cewnik centralny należy usunąć najszybciej jak to będzie możliwe.69,70
– W miarę możliwości nie należy podawać insuliny przez cewnik założony do żyły głównej, o ile nie jest to jedyna możliwa droga, ponieważ podawanie innych płynów przez tę samą linię centralną może prowadzić do przerwania wlewu insuliny.
• Chorym gorączkującym należy podać antybiotyki po wcześniejszym pobraniu odpowiedniego materiału na posiew z płynów ustrojowych.
• Cewnikowanie pęcherza moczowego zwykle nie jest konieczne, z wyjątkiem przypadków, kiedy dziecko jest nieprzytomne lub nie jest w stanie oddać moczu na żądanie (np. niemowlęta i ciężko chore małe dzieci).
Gdzie należy leczyć dziecko z DKA?
Dziecko z DKA należy leczyć na oddziale, na którym:
– pracuje doświadczony zespół pielęgniarski, przeszkolony w monitorowaniu i leczeniu DKA
– obowiązują pisemne wytyczne postępowania w DKA u dzieci
– jest dostęp do laboratorium w razie potrzeby częstych i szybkich oznaczeń parametrów biochemicznych.
We wszystkich możliwych przypadkach leczeniem w warunkach szpitalnych powinien kierować specjalista/pediatra (ew. diabetolog, endokrynolog lub anestezjolog – przyp. kons.) doświadczony i przeszkolony w zakresie terapii DKA. Jeśli ograniczenia geograficzne wymagają rozpoczęcia leczenia w mniej doświadczonym ośrodku o ograniczonych możliwościach terapeutycznych, należy zapewnić możliwość kontaktu telefonicznego lub organizacji telekonferencji z lekarzem doświadczonym w leczeniu DKA.
U dzieci z ciężką DKA (długotrwałe objawy, niewydolność krążenia lub obniżony poziom świadomości) lub narażonych na większe ryzyko obrzęku mózgu (np. wiek <5 lat, ciężka kwasica, niskie pCO2, duże stężenie mocznika we krwi) należy rozważyć natychmiastowe rozpoczęcie leczenia na oddziale intensywnej terapii (w miarę możliwoś ci pediatrycznym) lub na oddziale wyposażonym w taki sam sprzęt i zapewniającym stały nadzór (np. na oddziale diabetologii dziecięcej).17,71
Dzieci, u których cukrzycę rozpoznano jakiś czas temu, a ich rodziców przeszkolono w zakresie postępowania w przypadku chorób towarzyszących o ostrym przebiegu, hiperglikemii i ketozy, bez wymiotów lub ciężkiego odwodnienia, można leczyć w domu lub ambulatoryjnie (np. na szpitalnym oddziale ratunkowym), pod warunkiem że terapię nadzoruje doświadczony zespół diabetologiczny.25,72,73
Monitorowanie kliniczne i biochemiczne
Skuteczne leczenie DKA i HHS wymaga skrupulatnego monitorowania odpowiedzi klinicznej i biochemicznej w celu odpowiedniej oraz szybkiej modyfikacji terapii, w zależności od parametrów klinicznych lub laboratoryjnych (ramka 2.).
Na karcie obserwacji należy systematycznie (co godzinę) zapisywać objawy kliniczne, leki podawane doustnie i dożylnie, płyny oraz wyniki badań laboratoryjnych. Należy monitorować między innymi:
• co godzinę (lub częściej wg wskazań) parametry życiowe (częstotliwość rytmu serca, częstotliwość oddechów, ciśnienie tętnicze)
• co godzinę (lub częściej wg wskazań) objawy neurologiczne (skala Glasgow [tab. 3.]) pod kątem objawów ostrzegawczych obrzęku mózgu (p. niżej)
– ból głowy
– nieadekwatne zwolnienie częstotliwości rytmu serca
– nawrót wymiotów
– zmiana stanu neurologicznego (niepokój, drażliwość, nadmierna senność, nietrzymanie moczu i/lub stolca) lub swoiste objawy neurologiczne (np. porażenie nerwów czaszkowych, zaburzenia odruchu źrenicznego)
– podwyższenie ciśnienia tętniczego
– zmniejszone wysycenie hemoglobiny tlenem
– gwałtowne zwiększenie stężenia sodu w surowicy sugerujące utratę wolnej wody z moczem jako objaw moczówki prostej (będący efektem przerwania dopływu krwi do przysadki w następstwie wgłobienia mózgu)
• dawki insuliny
• co godzinę (lub częściej wg wskazań) dokładną objętość podawanych płynów (w tym wszystkich płynów przyjmowanych doustnie) oraz wydalanych
• co godzinę stężenie glukozy we krwi włośniczkowej (przy zaburzeniach perfuzji obwodowej i kwasicy metoda ta może być niedokładna, dlatego wynik należy zweryfikować w oznaczeniu stężenia glukozy we krwi żylnej)
• badania laboratoryjne: stężenie elektrolitów, glukozy, mocznika, wapnia, magnezu i fosforu w surowicy, hematokryt i gazometrię należy oznaczać co 2–4 godziny lub częściej u chorych z nasiloną DKA (w zależności od wskazań klinicznych) • jeśli jest taka możliwość, co 2 godziny stężenie BOHB we krwi53-57
– przyłóżkowe oznaczenia BOHB dobrze korelują z wynikami oznaczeń metodą referencyjną do wartości 3 mmol/l, ale są niedokładne, gdy stężenie BOHB przekracza 5 mmol/l55,74
• stężenie lipidów i trójglicerydów może być znacznie zwiększone, co w pobranej próbce krwi jest widoczne jako otoczka lipidowa75
• jeżeli laboratorium nie jest w stanie odpowiednio szybko podać wyników badań, ich przydatnym uzupełnieniem jest przenośny analizator biochemiczny, który pozwala na oznaczenie przy łóżku pacjenta stężenia elektrolitów w surowicy i gazometrii we krwi pobranej z opuszki palca; oczekując na wyniki oznaczeń wykonywanych w laboratorium, można już zbadać przyłóżkowo stężenie glukozy we krwi oraz związków ketonowych we krwi lub w moczu.
• przydatne obliczenia dodatkowe:
– luka anionowa = Na - (Cl + HCO3): norma 12±2 (mmol/l)
• w DKA luka anionowa zwykle wynosi 20–30 mmol/l; wynik >35 mmol/l sugeruje współistnienie kwasicy mleczanowej
– skorygowane stężenie sodu = oznaczone stężenie Na + 2 ([stężenie glukozy w osoczu-5,6]/5,6) mmol/l lub oznaczone stężenie Na + 2 ([stężenie glukozy w osoczu-100]/100) mg/dl
– efektywna osmolalność (mOsm/kg) = 2 × (stężenie Na w osoczu) + stężenie glukozy w osoczu (mmol/l).76
Płyny i elektrolity
U chorych z DKA występuje 5–10% niedobór płynu zewnątrzkomórkowego.8,9 U dzieci w przebiegu DKA rzadko występuje wstrząs z zaburzeniami hemodynamicznymi. Ocena kliniczna odwodnienia jest subiektywna i niedokładna,59-61 dlatego w przypadku umiarkowanej DKA należy przyjąć, że niedobór płynów wynosi 5–7%, a w ciężkiej kwasicy 7–10%. Efektywna osmolalność (obliczona wg wzoru podanego powyżej) wynosi zwykle 300–350 mOsm/kg. Zwiększone stężenie mocznika w surowicy oraz wartość hematokrytu lub stężenie hemoglobiny, a u chorych z podejrzeniem niedokrwistości stężenie albumin lub białka całkowitego w surowicy,77 mogą być pomocnymi wskaźnikami stopnia utraty płynu zewnątrzkomórkowego,73,78,79 dlatego należy je często oznaczać w trakcie resuscytacji płynowej i uzupełniania niedoboru.80 Stężenie sodu w surowicy nie jest natomiast wiarygodnym wskaźnikiem zmniejszenia objętości płynu zewnątrzkomórkowego z dwóch powodów: po pierwsze, glukoza, której większość znajduje się w przestrzeni zewnątrzkomórkowej, na skutek różnicy ciśnień osmotycznych powoduje przesunięcie wody do przestrzeni zewnątrzkomórkowej, a w rezultacie hiponatremię z rozcieńczenia,81,82 po drugie, w DKA dochodzi do zwiększenia frakcji lipidowej osocza, która zawiera mało sodu. To drugie zjawisko nie ma istotnego znaczenia w przypadku stosowaniu większości nowoczesnych metod oznaczania sodu. Przydatne jest obliczenie skorygowanego stężenia sodu (za pomocą wzoru podanego powyżej), które odpowiada oczekiwanemu stężeniu sodu w surowicy przy braku hiperglikemii, i monitorowanie jego zmian w trakcie leczenia kwasicy. Ponieważ stężenie glukozy w osoczu zmniejsza się po podaniu płynów i insuliny, oznaczone stężenie sodu powinno się zwiększyć, a skorygowane o stężenie glukozy stężenie sodu (obliczone na podstawie podanego powyżej wzoru) powinno się powoli zmniejszać. Należy jednak pamiętać, że zwiększenie oznaczonego stężenia sodu w surowicy nie oznacza nasilenia hiperosmolalności. Brak zwiększenia stężenia sodu lub dalsze pogłębianie się hiponatremii mimo stosowania leczenia uważa się za niekorzystny czynnik rokowniczy, sugerujący początek obrzęk mozgu.83-85 Zbyt szybkie i utrzymujące się zwiększenie stężenia sodu w surowicy także może wskazywać na możliwość obrzęku mózgu w wyniku utraty wolnej wody z moczem w następstwie moczówki prostej.
Celem terapii płynowej i elektrolitowej jest:
– przywrócenie prawidłowej objętości wewnątrznaczyniowej
– uzupełnienie sodu i niedoborów wody w przestrzeni zewnątrz- i wewnątrzkomórkowej
– poprawa filtracji kłębuszkowej wraz z poprawą usuwania glukozy i związków ketonowych z krwi.
Ramka 2. Cele leczenia DKA
wyrównanie odwodnienia
• wyrównanie kwasicy i odwrócenie ketozy
• przywrócenie stężenia glukozy we krwi do wartości zbliżonej do normy
• monitorowanie powikłań DKA i ich leczenie
• rozpoznanie i leczenie przyczyn rozwoju DKA
Zasady uzupełniania wody i elektrolitów
Pomimo wielu starań podejmowanych w celu poznania przyczyny obrzęku mózgu, jego patogeneza nadal nie jest w pełni jasna. Nie ma przekonujących danych potwierdzających związek pomiędzy tempem podawania płynów lub sodu w trakcie leczenia DKA a rozwojem obrzęku mozgu.86-88 Opierając się na wiarygodnych danych, nie można jednoznacznie wskazać, która strategia leczenia ma przewagę nad innymi metodami.87 Poniżej opisano zasady postępowania, które opracowano na podstawie szczegółowej analizy piśmiennictwa. Zostały one przyjęte i zatwierdzone przez grupę ekspertów reprezentujących Lawson Wilkins Pediatric Endocrine Society (LWPES), European Society for Paediatric Endocrinology (ESPE) i International Society for Pediatric and Adolescent Diabetes (ISPAD).17,89
• Uzupełnij niedobory wody i elektrolitów.
• Przy obliczaniu niedoboru oraz w trakcie leczenia uwzględnij płyny, które podano dożylnie lub doustnie w innym ośrodku.
• Resuscytacja płynowa. U pacjentów z ciężkim niedoborem płynów, lecz bez objawów wstrząsu, natychmiast rozpocznij intensywne wypełnianie łożyska naczyniowego (tzw. resuscytację płynową) 0,9% roztworem NaCl w celu przywrócenia krążenia obwodowego. Objętość podanych płynów zwykle wynosi 10–20 ml/kg mc. w ciągu 1–2 h, ale w celu uzyskania odpowiedniej perfuzji tkankowej konieczne może być powtórzenie tej dawki.
– W rzadkich przypadkach w przebiegu DKA dochodzi do wstrząsu. Należy wtedy jak najszybciej wyrównać objętość wewnątrznaczyniową, podając w bolusach przez kaniulę o dużej średnicy 0,9% roztwór NaCl w dawce 20 ml/kg mc. Po każdym bolusie należy ponownie ocenić stan pacjenta.
– Należy stosować roztwory krystaloidów, a nie koloidów. Brakuje danych wskazujących na przewagę koloidów nad krystaloidami w leczeniu DKA.
• Uzupełnienie niedoboru płynów. W dalszej terapii płynowej (uzupełnianie niedoboru) stosuj roztwory izotoniczne (0,9% roztwór NaCl, roztwór Ringera z dodatkiem mleczanu lub Plasmalyte) przez co najmniej 4–6 h.78,83,90-93
– U pacjentów z łagodną DKA zwykle nie do- chodzi do zaburzeń krążenia obwodowego, dlatego nie wymagają oni podawania płynów w bolusach. Terapię płynową rozpocznij od uzupełnienia niedoboru oraz wyrównania zapotrzebowania podstawowego.
• U wszystkich dzieci po zmniejszeniu stężenia glukozy we krwi w trakcie leczenia dochodzi do zmniejszenia objętości łożyska naczyniowego, dlatego niezwykle ważne jest zapewnienie wystarczającej ilości płynów i elektrolitów dla utrzymania odpowiedniej perfuzji tkankowej.
– Po 4–6 h niedobory płynowe uzupełniaj roztworem o ciśnieniu osmotycznym równo- ważnym lub większym od 0,45% roztworu NaCl z dodatkiem chlorku potasu, fosforanu potasu lub octanu potasu (p. zalecenia dotyczące uzupełniania potasu).78,83,90,94-96 Decyzja o zmianie roztworów izotonicznych na hipotoniczne zależy od stanu nawodnienia chorego, stężenia sodu w surowicy krwi i osmolalności.
– Poza podawaniem płynów w objętości pokrywającej zapotrzebowanie podstawowe równomiernie uzupełniaj obliczony niedobór płynów przez 48 h.17,78,97 Z wyjątkiem pacjentów w ciężkim stanie, doustne podawanie płynów zwykle rozpoczyna się w ciągu 24 h.97 Terapię płynową zaplanuj w taki sposób, aby prawidłowy stan nawodnienia przywrócić w ciągu 48 h. Jednak w badaniu obejmującym 635 pacjentów z DKA średni czas do skorygowania stanu DKA oraz całkowitego przywrócenia wydolności krążenia wynosił 11,6±6,2 h. Zaraz po ustąpieniu DKA pozostałe niedobory płynowe uzupełniano drogą doustną, a insulinę podawano podskórnie.97
– Ze względu na trudności w określeniu stopnia odwodnienia, który często jest zaniżony lub zawyżony,59-61 płyny należy podawać codziennie w tempie na ogół nieprzekraczającym 1,5–2-krotnego zwykłego dobowego zapotrzebowania, w zależności od wieku, masy lub powierzchni ciała.17 W tabeli 2. podano przykładowe obliczenia.
– Zadowalające wyniki uzyskano też po zastosowaniu alternatywnej, uproszczonej metody: po bolusie 0,9% roztworu NaCl w dawce 20 ml/kg mc. niezależnie od stopnia odwodnienia podaje się 0,675% roztwór NaCl (3/4 fizjologicznego roztworu soli, 115,5 mmol sodu) w objętości równej 2–2,5-krotności normalnego, podstawowego zapotrzebowania na płyny, następnie po 24 h lub wcześniej w przypadku ustąpienia kwasicy objętość podawanych płynów zmniejsza się do 1–1,5-krotności podstawowego zapotrzebowania, kontynuując takie postępowanie do czasu ustąpienia ketonurii.95,98
• Poza kliniczną oceną odwodnienia, w terapii płynowej i elektrolitowej pomocne może być obliczenie efektywnej osmolalności. Celem takiego postępowania jest stopniowe zmniejszenie efektywnej osmolalności osocza do wartości prawidłowej.80,97,99 Wraz ze zmniejszeniem stężenia glukozy w surowicy krwi należy jednocześnie doprowadzić do zwiększenia stężenia sodu (stężenie sodu powinno się zwiększać o 0,5 mmol/l na każdy 1 mmol/l zmniejszenia stężenia glukozy w surowicy).
• W obliczeniach objętości płynów do uzupełnienia zwykle nie trzeba uwzględniać wody utraconej z moczem, choć w wyjątkowych okolicznościach może być to konieczne.
• Należy zwiększyć zawartość sodu w podawanych płynach, jeżeli oznaczone stężenie sodu jest małe i nie zwiększa się odpowiednio do redukcji glikemii.83,93,99,100
• Podawanie dużej objętości płynu zawierającego znaczną ilość chloru (w powiązaniu z preferencyjnym wydalaniem związków ketonowych z moczem w porównaniu z chlorem) może bardzo szybko prowadzić do hiperchloremii101-103 (definiowanej jako stosunek chloru do sodu [Cl-:Na+] >0,79104) i hiperchloremicznej kwasicy metabolicznej.96,102,105-107
– W przypadku monitorowania poprawy biochemicznej na podstawie całkowitego niedoboru zasad działanie zakwaszające chloru może utrudniać rozpoznanie ustępowania DKA.103
– W trakcie hiperchloremii utrzymujący się niedobór zasad lub małe stężenie wodorowęglanu można błędnie zinterpretować jako wykładnik utrzymującej się kwasicy.
– Przyłóżkowy pomiar BOHB pozwala uniknąć błędnej interpretacji wyników, potwierdzając ustępowanie kwasicy. Kwasica hiperchloremiczna ustępuje samoistnie.
– Stosowanie oceny luki anionowej w celu monitorowania ustępowania kwasicy ma w tym przypadku dwa ograniczenia: nie pozwala odróżnić mieszanej kwasicy metabolicznej (ketonowej i hiperchloremicznej) oraz ilościowo określić stopnia kwasicy hiperchloremicznej.
• W prawidłowych warunkach różnica między stężeniami sodu a chloru w surowicy wynosi 30–35 mmol/l. Aby umożliwić lekarzom oddzie- lenie deficytu zasad związanego z obecnością chloru w trakcie przyłóżkowego monitorowania ustępowania DKA, zaproponowano następujący wzór: niedobór zasad wywołany chlorem = (stężenie sodu w osoczu – stężenie chloru w osoczu – 32).103
• Całkowitą ilość chloru można zmniejszyć, unikając podawania potasu w postaci chlorku potasu. Wskazane jest natomiast stosowanie roztworu Ringera z dodatkiem mleczanu lub Plasmalyte, w których chlor zastąpiono odpowiednio mleczanem lub octanem.108
Leczenie insuliną
Przyczyną DKA jest zmniejszenie efektywnego stężenia insuliny we krwi wraz ze zwiększeniem stężenia hormonów antagonistycznych. Samo nawodnienie w pewnym stopniu zmniejsza stężenie glukozy we krwi,109,110 jednak insulinoterapia jest niezbędna do przywrócenia prawidłowego metabolizmu komórek oraz do normalizacji glikemii i zahamowania lipolizy i ketogenezy.111
Liczne dane wskazują, że dożylne podawanie insuliny „w małej dawce” jest bezpieczne i skuteczne.97,98,112
• Wlew insuliny rozpocznij 1–2 h po wdrożeniu terapii płynowej (tj. po wstępnym uzupełnieniu niedoboru płynów).88
• Wyrównanie niedoboru insuliny:
– dawka: 0,05–0,1 j./kg mc./h (jedną z metod jest rozpuszczenie 50 j. insuliny krótko działającej w 50 ml fizjologicznego roztworu soli, 1 j. = 1 ml)113-120
– droga podania: dożylnie
– na początku leczenia nie podawaj bolusów dożylnie; nie jest to konieczne,119,121a zwiększa ryzyko obrzęku mozgu88,99,122 i może nasilić hipokaliemię.
• Dawkę insuliny zwykle utrzymuj na poziomie 0,05–0,1 j./kg mc./h co najmniej do momentu ustąpienia DKA (pH >7,30, stężenie wodorowęglanu >15 mmol/l, BOHB <1 mmol/l lub luka anionowa w zakresie normy), co zazwyczaj trwa dłużej niż normalizacja glikemii.123
• Jeżeli u pacjenta stwierdza się wyraźną insulino wrażliwość (np. u małych dzieci z DKA, pacjentów z HHS i u niektórych starszych dzieci chorujących na cukrzycę od dłuższego czasu), dawkę insuliny można zmniejszyć, pod warunkiem że nie zahamuje to ustępowania kwasicy metabolicznej. Na przykład zapobieganie hipoglikemii u małego dziecka otrzymującego insulinę w dawce 0,05 j./kg mc./h może wymagać jej zmniejszenia do 0,03 j./kg mc./h.
• W badaniach z retrospektywnym zbieraniem danych bez grupy kontrolnej oraz w badaniach obserwacyjnych wykazano podobną skuteczność i bezpieczeństwo dawki 0,05 j./kg mc./h,124,125 a w niektórych ośrodkach pediatrycznych taką dawkę stosuje się rutynowo w leczeniu DKA. Dotychczas nie przeprowadzono dobrze zaplanowanych badań klinicznych z randomizacją, jednak z drugiej strony nie ma danych wskazujących na niekorzystne działanie większej dawki insuliny.
• Insulina wykazuje działanie podobne do aldosteronu, zwiększając wydalanie potasu z moczem.126-130 Insulina podawana dożylnie w dużej dawce przez dłuższy czas może się przyczynić do zmniejszenia stężenia potasu w surowicy w następstwie zwiększonego wydalania z moczem, pomimo dodatkowego podawania potasu.
– W celu uniknięcia ciężkiej hipokaliemii należy ograniczać czas podawania insuliny dożylnie oraz zmniejszać jej dawkę.131
• Podczas wstępnego wyrównywania niedoboru płynów stężenie glukozy w osoczu gwałtownie się zmniejsza.109 Następnie, po rozpoczęciu insulinoterapii, stężenie zwykle zmniejsza się w tempie 2–5 mmol/l/h, w zależności od czasu podawania glukozy i jej dawki.113-116,118,119,132
• W celu zapobiegania zbyt szybkiemu zmniejszeniu się stężenia glukozy w osoczu i hipoglikemii, do płynów podawanych dożylnie należy dodać 5% roztwór glukozy (np. 5% glukozę do 0,45% roztworu NaCl), gdy stężenie glukozy zmniejszy się do około 250–300 mg/dl (14–17 mmol/l) lub wcześniej, jeśli glikemia zmienia się bardzo szybko.
– Konieczne może być podanie 10% lub nawet 12,5% roztworu glukozy w celu uniknięcia hipoglikemii przy utrzymaniu wlewu insuliny mającego na celu wyrównanie kwasicy metabolicznej.
• Jeżeli stężenie glukozy zmniejsza się bardzo szybko >88 mg/dl/h (5 mmol/l/h) po wstępnym uzupełnieniu niedoboru płynów, należy rozważyć dodanie glukozy do podawanych płynów jeszcze zanim jej stężenie w osoczu zmniejszy się do 300 mg/dl (17 mmol/l).
• W przypadku braku poprawy wskaźników biochemicznych kwasicy (pH, luka anionowa, stężenie BOHB), należy ponownie ocenić stan pacjenta i dawkowanie insuliny oraz rozważyć inne możliwe przyczyny osłabionej odpowiedzi na insulinę, na przykład zakażenie lub błędy w przygotowaniu wlewu insuliny.
• Jeśli podawanie insuliny w ciągłym wlewie dożylnym jest niemożliwe oraz u chorych z niepowikłaną DKA, bezpieczne jest podskórne lub domięśniowe wstrzyknięcie krótko lub szybko działającego analogu insuliny (insulina lispro lub insulina aspart) powtarzane co 1–2 h. Takie postępowanie może być równie skuteczne jak dożylny wlew insuliny krótko działającej,132-136 lecz metody tej nie należy stosować u chorych z zaburzeniami krążenia obwodowego.
– Dawka początkowa s.c.: 0,3 j./kg mc., a następnie po 1 h insulina lispro lub aspart w dawce 0,1 j./kg mc. s.c. co godzinę lub 0,15–0,20 j./kg mc. co 2 h.
– Jeśli stężenie glukozy zmniejszy się do wartości <250 mg/dl (14 mmol/l) przed ustąpieniem DK, należy zmniejszać dawkę insuliny lispro lub aspart do 0,05 j./kg mc. s.c. co godzinę w celu utrzymania glikemii na poziomie około 200 mg/dl (11 mmol/l) do czasu ustąpienia DKA.
Uzupełnianie potasu
U dzieci z DKA występuje niedobór całkowitej zawartości potasu w organizmie rzędu 3–6 mmol/kg mc.8-12 Najwięcej utraconego potasu pochodzi z przestrzeni wewnątrzkomórkowej. Przyczyną wewnątrzkomórkowego niedoboru potasu jest przesunięcie tego jonu przez błonę komórkową na skutek zwiększonego ciśnienia osmotycznego (zwiększona osmolalność osocza powoduje przesunięcie wody i potasu na zewnątrz komórki), a będąca efektem niedoboru insuliny glikogenoliza i proteoliza powoduje wypływ potasu z komórek. Pacjent traci też potas wskutek wymiotów oraz diurezy osmotycznej. Niedobór płynu wewnątrznaczyniowego jest przyczyną wtórnego hiperaldosteronizmu, który nasila wydalanie potasu z moczem. W efekcie dochodzi do niedoboru całkowitej zawartości potasu w organizmie, lecz początkowo stężenie potasu w surowicy może być prawidłowe, zwiększone lub zmniejszone.137 Upośledzenie czynności nerek przyczynia się do hiperkaliemii poprzez nasilenie hiperglikemii i zmniejszenie wydalania potasu.137 Podanie insuliny i wyrównanie kwasicy ułatwia przesunięcie potasu z powrotem do wnętrza komórek, zmniejszając jego stężenie w surowicy.138 To zmniejszenie może nastąpić nagle, predysponując do wystąpienia zaburzeń rytmu serca.
Uzupełnienie potasu jest konieczne bez względu na jego stężenie w osoczu, z wyjątkiem pacjentów z niewydolnością nerek.139,140
• W przypadku hipokaliemii suplementację potasu należy rozpocząć jednocześnie ze wstępną terapią płynową i przed wdrożeniem insulinoterapii. W pozostałych przypadkach suplementację należy rozpocząć po wstępnym wyrównaniu niedoboru płynów i równocześnie z insulinoterapią. Hiperkaliemia jest wskazaniem do opóźnienia uzupełniania potasu do czasu potwierdzenia prawidłowej diurezy.
• Jeżeli niezwłoczne oznaczenie stężenia potasu w surowicy jest niemożliwe, wykonanie zapisu EKG może pomóc w ustaleniu, czy u dziecka występuje hiper- czy hipokaliemia.65,66 Wydłużenie odstępu PR, spłaszczenie i odwrócenie załamka T, obniżenie odcinka ST, pojawienie się załamka U i widoczne wydłużenie odstępu QT (wynikające z połączenia załamków T i U) wskazują na hipokaliemię. Wysokie, szpiczaste i symetryczne załamki T oraz skrócenie odstępu QT są objawami hiperkaliemii.
• Początkowe stężenie potasu we wlewie kroplowym powinno wynosić 40 mmol/l. Dalszą suplementację należy prowadzić na podstawie wyniku pomiarów stężenia potasu w surowicy.
– Jeżeli potas podaje się równocześnie z szybkim uzupełnieniem niedoboru płynów, jego stężenie powinno wynosić 20 mmol/l.
• Fosforan potasu można stosować wraz z chlorkiem lub octanem potasu, na przykład 20 mmol/l chlorku potasu i 20 mmol/l fosforanu potasu lub 20 mmol/l fosforanu potasu i 20 mmol/l octanu potasu. Podawanie potasu wyłącznie w postaci chlorku potasu zwiększa ryzyko metabolicznej kwasicy hiperchloremicznej, natomiast podawanie wyłącznie fosforanu potasu może prowadzić do hipokalcemii.
• Uzupełnianie potasu należy kontynuować przez cały okres dożylnego wyrównywania niedoborów płynów.
• Maksymalne zalecane tempo dożylnego podawania potasu wynosi zwykle 0,5 mmol/kg mc./h.
• Jeżeli hipokaliemia utrzymuje się mimo maksymalnego tempa wlewu potasu, można zmniejszyć tempo wlewu insuliny.
Fosforany
W DKA dochodzi do wewnątrzkomórkowego niedoboru fosforanów na skutek diurezy osmotycznej8-10Stężenie fosforanów w osoczu zmniejsza się po rozpoczęciu leczenia, a insulina nasila ten proces, ułatwiając wnikanie fosforanów do wnętrza komórek.141-143 Całkowity niedobór fosforanów w organizmie prowadzi do różnych zaburzeń metabolicznych.144-146 Klinicznie istotna hipofosfatemia może wystąpić podczas dożylnego podawania płynów przez ponad 24 h bez żywienia drogą doustną.8-10
• W badaniach z prospektywnym zbieraniem danych, obejmujących stosunkowo małą liczbę pacjentów (a w wyniku tego o ograniczonej mocy statystycznej), nie wykazano korzyści klinicznych z uzupełniania fosforanów.147-152
• Ciężka hipofosfatemia w skojarzeniu z niedoborem fosforanów (tzn. niebędąca jedynie wynikiem przesunięcia fosforanów do przestrzeni wewnątrzkomórkowej) występuje rzadko, ale może mieć bardzo poważne następstwa. Obraz kliniczny zależy od nasilenia i czasu utrzymywania się niedoboru fosforanów, a objawy kliniczne zwykle występują po zmniejszeniu stę- żenia fosforanów w osoczu do wartości <1 mg/dl (0,32 mmol/l).
• Ciężka hipofosfatemia może wystąpić w trakcie leczenia DKA, jednak jej objawy kliniczne występują rzadko, ponieważ zmniejszenie stężenia fosforanów w osoczu zwykle ma charakter ostry i nie poprzedza go przewlekły niedobór tych związków.
• Objawy kliniczne hipofosfatemii są głównie wynikiem zmniejszenia ilości fosforanów wewnątrz komórek. Zmniejszenie poziomu wewnątrzkomórkowego ATP upośledza czynność komórek zależną od wysokoenergetycznych związków fosforanowych, a zmniejszenie poziomu 2,3-difosfoglicerolu (DPG) zwiększa powinowactwo hemoglobiny do tlenu, ograniczając uwalnianie tlenu w tkankach.152 Dotyczy to wielu narządów i układów.145,153 Objawy kliniczne obejmują:
– encefalopatię metaboliczną (drażliwość, parestezje, splątanie, drgawki, śpiączkę); upośledzenie kurczliwości mięśnia sercowego i niewydolność oddechową z powodu osłabienia przepony; zaburzenia czynności mięśni z osłabieniem mięśni proksymalnych, zaburzenia połykania i niedrożność jelit; rzadko objawy ze strony układu krwiotwórczego obejmujące hemolizę, osłabienie fagocytozy i chemotaksji granulocytów, upośledzenie rozpuszczania skrzeplin (fibrynolizy) oraz małopłytkowość. Ostra hipofosfatemia u pacjentów z wcześniejszym ciężkim niedoborem fosforanów może prowadzić do rozpadu mięśni szkieletowych (rabdomiolizy).145,154,155
• Ciężka hipofosfatemia i współwystępowanie któregokolwiek z wymienionych powyżej objawów wymaga leczenia.1560157
• Podawanie fosforanów może wywołać hipokalcemię.158,159
• Fosforan potasu można bezpiecznie stosować jako alternatywę lub w skojarzeniu z chlorkiem albo octanem potasu, jednak konieczne jest ścisłe monitorowanie stężenia wapnia w surowicy w celu uniknięcia hipokalcemii.158,159
Ramka 3. Powikłania leczenia DKA
• niedostateczne uzupełnienie niedoboru płynów
• hipoglikemia
• hipokaliemia
• kwasica hiperchloremiczna
• obrzęk mózgu
Kwasica
Ciężką kwasicę można odwrócić poprzez podawanie płynów i insuliny. Insulina hamuje dalsze wytwarzanie ketokwasów i umożliwia ich metabolizm, w wyniku czego powstaje wodorowęglan. Leczenie hipowolemii poprawia perfuzję tkanek i czynność nerek, zwiększając w ten sposób wydalanie kwasów organicznych.
Badania z grupą kontrolną nie wykazały klinicznych korzyści z podawania wodorowęglanu.160-163Wodorowęglan może wywoływać paradoksalną kwasicę ośrodkowego układu nerwowego (OUN),164,165 a szybkie wyrównywanie kwasicy za pomocą wodorowęglanu prowadzi do hipokaliemii.164,166,167 Podawanie wodorowęglanu może natomiast być korzystne w rzadkich przypadkach zagrażającej życiu hiperkaliemii.168
• Jeżeli zastosowanie wodorowęglanu uzna się za konieczne, należy go podać ostrożnie w dawce 1–2 mmol/kg mc. w ciągu 60 minut (E). Powikłania leczenia DKA wymieniono w ramce 3.
Doustne podawanie płynów i przejście na podawanie insuliny podskórnie
• Doustne podawanie płynów rozpoczynaj tylko w przypadku istotnej poprawy klinicznej (obecna może być nadal lekka kwasica/ketoza).
– Charakterystyczne jest utrzymywanie się ketonurii (oznaczenie obecności związków ketonowych w moczu za pomocą testu pasko- wego opiera się na reakcji z nitroprusydkiem, wykrywającej acetooctan i aceton) jeszcze przez kilka godzin po powrocie stężenia BOHB w surowicy do normy.53,57
– Brak związków ketonowych w moczu nie jest dowodem ustąpienia DKA.
• Po uzyskaniu dobrej tolerancji płynów doustnych zmniejsz objętość płynów podawanych dożylnie, tak aby łączna objętość płynów podawanych dożylnie i doustnie nie przekraczała obliczonego zapotrzebowania na płyny i.v. (tzn. nie przekraczała 1,5–2-krotności zapotrzebowania podstawowego). Takie ograniczenie płynowe należy stosować przez 48 h po przyjęciu do szpitala (w przypadku ciężkiej hiperosmolalności w początkowej fazie leczenia okres ten powinien wynosić 72 h).
• Po ustąpieniu DKA i uzyskaniu tolerancji żywienia doustnego zaplanuj zmianę drogi podawania insuliny na podskórną (s.c.). Najdogodniejszym momentem na wprowadzenie tej zmiany jest okres tuż przed posiłkiem.
• W celu profilaktyki hiperglikemii „z odbicia”, pierwszą dawkę insuliny s.c. należy podać 15–30 minut (dla szybko działającego analogu) lub 1–2 h (dla insuliny krótko działającej) przed zatrzymaniem jej wlewu. Takie postępowanie umożliwia wchłonięcie się insuliny podanej s.c. W przypadku insulin o pośrednim i długim czasie działania należy dłużej stosować insulinę we wlewie, stopniowo zmniejszając jej dawkę. Na przykład u pacjentów leczonych według schematu intensywnej insulinoterapii pierwszą dawkę insuliny podstawowej (bazowej) można podać wieczorem, a wlew dożylny zatrzymać następnego dnia rano.
• Dawka i rodzaj insuliny podawanej s.c. powinny być takie, jak zwykle stosuje się w danym ośrodku.
• Po przejściu na podawanie insuliny podskórnie konieczne jest częste monitorowanie glikemii w celu uniknięcia znacznej hiper- i hipoglikemii.
Powikłania i śmiertelność
Śmiertelność z powodu DKA u dzieci w badaniach populacyjnych wynosi 0,15–0,30%169-171 i może się zmniejszać.171 Główną przyczyną powikłań i zgonu jest uszkodzenie mózgu.170,172 Obrzęk mózgu stanowi przyczynę 60–90% wszystkich zgonów w przebiegu DKA.85,173 U 10–25% osób, które przeżyły epizod obrzęku mózgu, stwierdza się poważne trwałe następstwa.85,173,174 Dzieci bez widocznych objawów neurologicznych w trakcie leczenia DKA mogą po ustąpieniu kwasicy wykazywać subtelne objawy uszkodzenia mózgu, zwłaszcza zaburzenia pamięci.175
Inne rzadkie powikłania i przyczyny zgonu obejmują:
– hipokaliemięb
– hipokalcemię, hipomagnezemię
– ciężką hipofosfatemię
– hipoglikemię
– inne powikłania ze strony OUN (np. zakrzepicę zatok żylnych opony twardej, zakrzepicę tętnicy podstawnej, krwawienie śródczaszkowe i udar niedokrwienny)176-178
– zakrzepicę żył obwodowych69,70b
– zatorowość płucnąb
– posocznicę
– mukormykozę nosowo-mózgową lub płucną179
– zachłystowe zapalenie płucb
– obrzęk płucb
– zespół ostrej niewydolności oddechowej dorosłych (acute respiratory distress syndrome – ARDS)
– odmę opłucnową, odmę śródpiersia i odmę podskórną180
– rozpad mięśni szkieletowychb
– martwicę niedokrwienną jelit
– ostrą niewydolność nerekb
– ostre zapalenie trzustki.181,b
Obrzęk mózgu
Według badań populacyjnych prowadzonych w różnych krajach obrzęk mózgu rozwija się u 0,5–0,9% chorych, a związana z nim śmiertelność sięga 21–24%.85,173,174 U około 15% dzieci leczonych z powodu DKA występują jednak zaburzenia psychiczne (punktacja w skali Glasgow <14) towarzyszące objawom obrzęku mózgu w badaniach obrazowych.182,183 U pacjentów po okresie młodzieńczym powikłanie to obserwuje się rzadko. Wyniki badań obrazowych przekonują, że obrzęk mózgu nie jest rzadkim zjawiskiem u dzieci z DKA – występuje często z różnym nasileniem.182,184,185 Objawowy obrzęk mózgu jest prawdopodobnie najbardziej zaawansowaną postacią tego częstego powikłania.186
Przyczyna obrzęku mózgu jest niejasna. Według niektórych hipotez patogeneza obrzęku mózgu wiąże się z bardzo szybkim podaniem płynów, prowadzącym do gwałtownej zmiany osmolalności surowicy.100,187-190 Wyniki nowszych badań wskazują jednak, że odwodnienie i zmniejszony przepływ krwi przez naczynia mózgowe mogą się wiązać z uszkodzeniem mózgu w przebiegu DKA.85,191-193
Obserwacje te skłoniły badaczy do postawienia przeciwnej hipotezy, według której przyczyną uszkodzenia mózgu są czynniki związane z samą DKA, nasilające się w trakcie leczenia.194,195 Warto odnotować, że stopień ciężkości obrzęku mózgu w przebiegu DKA koreluje z wyjściowym stopniem odwodnienia i hiperwentylacji, a nie z czynnikami związanymi z wyjściową osmolalnością lub zmianami ciśnienia osmotycznego w trakcie leczenia.183 U dzieci zmarłych z powodu obrzęku mózgu w przebiegu DKA wykazano przerwanie bariery krew–mózg,196,197 co potwierdza pogląd, że obrzęk mózgu nie jest prostą konsekwencją zmniejszenia osmolalności surowicy.
Do czynników demograficznych zwiększających ryzyko obrzęku mózgu należą między innymi:
– młodszy wiek198
– nowo rozpoznana cukrzyca170,198
– dłuższy czas trwania objawów.199
Powyższe czynniki ryzyka mogą odzwierciedlać większe prawdopodobieństwo ciężkiej DKA.
W badaniach epidemiologicznych określono szereg potencjalnych czynników ryzyka stwierdzanych w momencie rozpoznania lub w trakcie leczenia DKA. Należą do nich:
– większa hipokapnia (po uwzględnieniu poprawki na nasilenie kwasicy)85,183,200
– większe wyjściowe stężenie mocznika w surowicy85,183
– większe wyjściowe nasilenie kwasicy88,201,202
– stosowanie wodorowęglanu w celu wyrównywania kwasicy85,203
– znaczne wczesne zmniejszenie efektywnej osmolalności surowicy99,202
– wolniejsze zwiększenie stężenia sodu w surowicy lub wczesne zmniejszenie stężenia sodu skorygowanego o stężenie glukozy w trakcie leczenia83-85,202
– większa objętość płynów podawanych w ciągu pierwszych 4 godzin88,200,202
– podawanie insuliny w pierwszej godzinie wyrównywania niedoboru płynów.88
Do objawowego obrzęku mózgu zwykle dochodzi w ciągu pierwszych 12 h po rozpoczęciu leczenia, ale także może on wystąpić przed rozpoczęciem leczenia85,174,204-207 lub w rzadkich przypadkach po 24–48 h.85,198,208 Objawy przedmiotowe i podmiotowe obrzęku mózgu są zmienne (ramka 4.). Łagodny i umiarkowany ból głowy może występować stosunkowo często w chwili rozpoznania (informacja uzyskana osobiście przez Nicole Glaser), jednak silny ból głowy po rozpoczęciu leczenia jest zawsze niepokojący. Poniżej opisano metodę rozpoznania klinicznego na podstawie oceny stanu neurologicznego przy łóżku chorego.209 W przypadku stwierdzenia jednego kryterium diagnostycznego, dwóch kryteriów większych lub jednego większego i dwóch mniejszych czułość rozpoznania
wynosi 92%, a odsetek rozpoznań fałszywie dodatnich jedynie 4%. W rozpoznaniu obrzęku mózgu nie należy uwzględniać objawów stwierdzanych przed rozpoczęciem leczenia.
• Kryteria diagnostyczne
– nieprawidłowa odpowiedź ruchowa lub słowna na ból
– pozycja odkorowana lub odmóżdżeniowa
– porażenie nerwów czaszkowych (zwłaszcza III, IV i VI)
– nieprawidłowy neurogenny rytm oddychania (np. postękiwanie, tachypnöe, oddech Cheyne’a i Stokesa, bezdech).
• Kryteria większe
– zmiany stanu psychicznego/zmienny poziom przytomności
– utrzymujące się zmniejszenie częstotliwości rytmu serca (o >20 uderzeń/min), niewynikające z wyrównania objętości wewnątrznaczyniowej lub snu
– brak kontroli czynności fizjologicznych (adekwatnej dla wieku).
• Kryteria mniejsze
– wymioty
– ból głowy
– apatia lub trudności z wybudzeniem
– rozkurczowe ciśnienie tętnicze >90 mm Hg
– wiek <5 lat.
W dokumentacji lub przy łóżku chorego powinna być łatwo dostępna tabela z zakresami wartości referencyjnych ciśnienia tętniczego i częstotliwości rytmu serca w zależności od wysokości i masy ciała oraz płci.
Wystąpienie objawów moczówki prostej w postaci zwiększonej objętości moczu z jednoczesnym istotnym zwiększeniem stężenia sodu w surowicy, związanych z utratą wolnej wody z moczem, jest efektem wgłobienia mózgu prowadzącego do przerwania dopływu krwi do przysadki.
Ramka 4. Przedmiotowe i podmiotowe objawy ostrzegawcze obrzęku mózgu
• ból głowy i zmniejszenie częstotliwości rytmu serca
• zmiana stanu neurologicznego (niepokój, drażliwość, nasilona senność, nietrzymanie moczu i/lub stolca)
• swoiste objawy neurologiczne (np. porażenie nerwów czaszkowych, obrzęk tarczy nerwu wzrokowego)
• podwyższenie ciśnienia tętniczego
• zmniejszenie wysycenia hemoglobiny tlenem
Leczenie obrzęku mózgu
• W przypadku podejrzenia obrzęku mózgu leczenie rozpocznij natychmiast.
• Tempo podawania płynów zmniejsz o 1/3.
• W leczeniu stosuje się mannitol w dawce 0,5–1 g/kg mc. i.v. przez 10–15 minut. Powtórz
dawkę, jeśli odpowiedź na leczenie nie nastąpi w ciągu 30 minut do 2 godzin.210-212
• Alternatywnie do mannitolu można zastosować hipertoniczny (3%) roztwór NaCl w dawce 2,5–5 ml/kg mc. i.v. przez 10–15 minut, zwłaszcza w razie braku wstępnej odpowiedzi na mannitol.213,214
– W niedawno przeprowadzonym retrospektywnym badaniu kohortowym z 11-letnim okresem obserwacji wykazano, że w wielu ośrodkach amerykańskich hipertoniczny roztwór NaCl zastąpił mannitol i jest on najczęściej stosowanym roztworem hiperosmolalnym. Dostępne dane sugerują jednak, że zastosowanie hipertonicznego roztworu NaCl może nie mieć przewagi nad mannitolem i wiązać się ze zwiększeniem śmiertelności. Hipotezy te trzeba jednak potwierdzić w dodatkowych badaniach.171
• Płyny hipertoniczne powinny być łatwo dostępne przy łóżku pacjenta.
• Unieś łóżko po stronie głowy pacjenta o 30°.
• U chorych z postępującą niewydolnością oddechową konieczna może być intubacja.
• Po rozpoczęciu leczenia obrzęku mózgu należy rozważyć wykonanie badania obrazowego OUN, podobnie jak w przypadku chorych w bardzo ciężkim stanie z encefalopatią lub ostrymi objawami neurologicznymi o charakterze ogniskowym. Głównym celem badania jest wykluczenie zmian wymagających natychmiastowej operacji neurochirurgicznej (np. krwawienia wewnątrzczaszkowego) lub leczenia przeciwkrzepliwego (np. zakrzepica naczyń mózgu).177,215-217
Stan hiperglikemiczno-hiperosmolalny
HHS charakteryzuje się skrajnie dużym stężeniem glukozy w surowicy i hiperosmolalnością bez istotnej ketozy.32 HHS występuje coraz częściej,27,28,218 niemniej jednak u dzieci jest rzadszy niż DKA.
W przeciwieństwie do typowych objawów DKA (hiperwentylacja, wymioty i ból brzucha), które zwykle są przyczyną zgłaszania się do lekarza, stopniowo zwiększający się wielomocz i nadmierne pragnienie związane z HHS mogą przebiegać nie zauważenie, prowadząc do znacznego odwodnienia i utraty elektrolitów. Szacuje się, że w przebiegu HHS dorośli tracą 2-krotnie więcej płynów niż w trakcie DKA, ponadto ocenę stanu odwodnienia może utrudniać otyłość i hiperosmolalność. Pomimo znacznej utraty płynów i elektrolitów, hipertoniczność zapobiega zmniejszeniu objętości płynu wewnątrznaczyniowego, w związku z czym objawy odwodnienia mogą być mniej widoczne.
Zmniejszenie osmolalności w trakcie leczenia (w następstwie zwiększonego wydalania glukozy w moczu i stymulowanego insuliną przechodzenia glukozy do komórek) powoduje przesunięcie płynu wewnątrznaczyniowego i zmniejszenie objętości wewnątrznaczyniowej, a u chorych ze skrajnie dużym stężeniem glukozy w osoczu diureza osmotyczna może się utrzymywać przez wiele godzin. W początkowym okresie leczenia utrata płynów z moczem może być bardzo duża. Uzupełnianie niedoborów płynowych u chorych z HHS należy prowadzić intensywniej niż u dzieci z DKA, ponieważ w trakcie leczenia HHS może dojść do gwałtownego zmniejszenia objętości płynu w łożysku naczyniowym.
Leczenie HHS
Brakuje prospektywnie zebranych danych pozwalających na opracowanie wytycznych dotyczących leczenia HHS. Podane zalecenia oparto na rozległym doświadczeniu w leczeniu dorosłych oraz różnicach w patofizjologii HHS i DKA (p. ryc. 3.).32 Chorych należy hospitalizować na oddziale intensywnej terapii lub na oddziale o podobnym poziomie opieki, dysponującym specjalistyczną opieką medyczną, pielęgniarską oraz zapleczem laboratoryjnym.
Terapia płynowa
Celem początkowego leczenia jest zwiększenie objętości płynu w przestrzeni wewnątrz- i zewnątrznaczyniowej oraz przywrócenie prawidłowego przepływu krwi przez nerki. Niedobór płynów należy uzupełniać szybciej niż u chorych z DKA.
• Początkowo podaj bolus 0,9% NaCl w dawce ≥20 ml/kg mc. Przyjmij, że niedobór płynów wynosi około 12–15% masy ciała. W zależności od potrzeby podaj dodatkowe bolusy płynów w celu przywrócenia obwodowego przepływu krwi.
• W celu dalszego uzupełniania niedoboru płynów podawaj 0,45–0,75% roztwór NaCl w ciągu 24–48 h.
• Celem leczenia jest stopniowe zmniejszanie stężenia sodu w surowicy i osmolalności.
• Roztwory izotoniczne skuteczniej utrzymują objętość wewnątrznaczyniową, dlatego w przypadku niewystarczającej perfuzji i niewydolności hemodynamicznej w następstwie zmniejszenia osmolalności surowicy ponownie rozpocznij podawanie izotonicznego roztworu NaCl.
• Często mierz stężenie sodu w surowicy. Jego stężenie w podawanych płynach należy tak dostosować, aby ułatwiać stopniowe zmniejszanie skorygowanego stężenia sodu w surowicy. Zanotowane zgony były związane z nieskutecznym zmniejszaniem skorygowanego stężenia sodu w surowicy w trakcie leczenia, co może być wskazaniem do hemodializy. Odsetek przeżyć wśród chorych poddanych hemodializie wyniósł 80%, a w grupie objętej dializą otrzewnową – 20%.28
– Brakuje danych określających optymalne tempo zmniejszania stężenia sodu w surowicy. Zaleca się, aby u chorych w stanie odwodnienia hipernatremicznego wynosiło ono 0,5 mmol/l/h.219 Samo odpowiednie nawadnianie (tzn. przed rozpoczęciem insulinoterapii) powinno prowadzić do zmniejszenia stężenia glukozy w surowicy o 75–100 mg/dl/h (4,1–5,5 mmol/l/h).220,221
– Stężenie glukozy w surowicy zwykle szybciej zmniejsza się w kilku pierwszych godzinach leczenia, kiedy w związku ze zwiększeniem objętości płynu w łożysku naczyniowym poprawia się przepływ krwi przez nerki. Jeśli po kilku pierwszych godzinach stężenie glukozy w surowicy nadal szybko się zmniejsza (>90 mg/dl, 5 mmol/l/h), rozważ dodanie do płynów nawadniających 2,5 lub 5% roztworu glukozy. Brak oczekiwanego zmniejszenia stężenia glukozy w surowicy wymaga natychmiastowej ponownej analizy klinicznej z oceną czynności nerek.
– W przeciwieństwie do DKA, w leczeniu HHS zaleca się uzupełnianie sodu wydalanego z moczem.120 Stężenie sodu w moczu w trakcie diurezy osmotycznej odpowiada mniej więcej 0,45% NaCl, jednak w przypadku wątpliwości co do odpowiedniej objętości płynu w łożysku naczyniowym straty można wyrównywać płynami zawierającymi większe stężenie sodu.
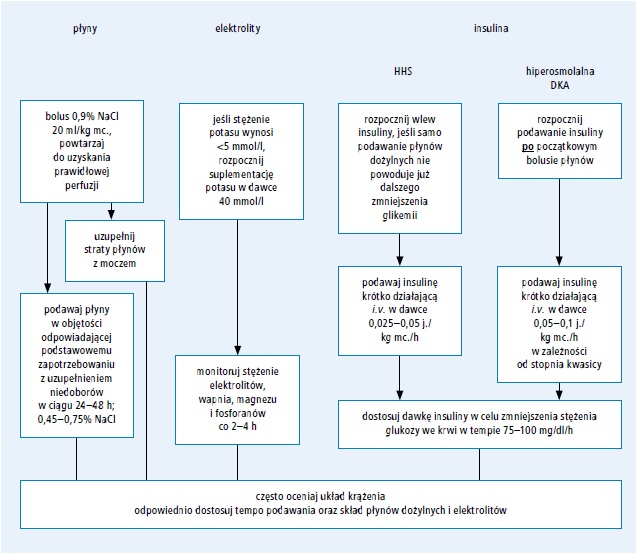
Ryc. 3. Leczenie stanu hiperglikemiczno-hiperosmolalnego (opracowano na podstawie 32. pozycji piśmiennictwa) Skróty: DKA – cukrzycowa kwasica ketonowa, HHS – stan hiperglikemiczno-hiperosmolalny
Leczenie insuliną
Zmniejszenie perfuzji tkanek w przebiegu HHS często prowadzi do kwasicy mleczanowej, natomiast kwasica ketonowa jest zwykle minimalna. W leczeniu HHS konieczne jest wczesne podanie insuliny. Już samo podawanie płynów znacznie zmniejsza stężenie glukozy w surowicy w wyniku rozcieńczenia, poprawia przepływ krwi przez nerki, nasilając glukozurię, a poprawa wydolności krążenia przyczynia się do zwiększonego zużycia glukozy w tkankach. Ciśnienie osmotyczne wywierane przez glukozę w łożysku naczyniowym pozwala utrzymać objętość krwi krążącej. Gwałtowne zmniejszenie stężenia glukozy w surowicy i osmolalności po podaniu insuliny w przypadku niedostatecznego uzupełnienia płynów może prowadzić do niewydolności krążenia i zakrzepicy. U pacjentów z HHS stwierdza się również skrajny niedobór potasu, a gwałtowne przesunięcie potasu do przestrzeni wewnątrzkomórkowej w wyniku działania insuliny może wywołać zaburzenia rytmu serca.
• Podawanie insuliny należy rozpocząć, jeśli w trakcie wlewu samych płynów tempo zmniejszania stężenia glukozy w surowicy nie przekracza już 50 mg/dl/h (3 mmol/l/h).
• U chorych z ketozą i kwasicą cięższego stopnia podawanie insuliny rozpocznij wcześniej.
• Początkowo insulinę można podać we wlewie ciągłym w dawce 0,025–0,05 j./kg mc./h. Dawkę insuliny należy dostosować w taki sposób, aby stężenie glukozy w surowicy zmniejszało się w tempie 50–75 mg/dl/h (3–4 mmol/l/h).
– Nie zaleca się podawania insuliny w bolusie.
Elektrolity
U chorych z HHS na ogół stwierdza się większy niedobór potasu, fosforanów i magnezu niż u chorych z DKA.
• Uzupełnianie potasu (40 mmol/l podawanych płynów) rozpocznij, gdy stężenie potasu mieści się w granicach normy, a czynność nerek jest prawidłowa.
– Po rozpoczęciu wlewu insuliny konieczne może być szybsze uzupełnianie potasu.
– Stężenie potasu w surowicy wraz z zapisem EKG należy kontrolować co 2–3 h.
– U chorych z hipokaliemią konieczne może być kontrolowanie stężenia potasu co godzinę.
• Podawanie wodorowęglanu jest przeciwskazane, ponieważ zwiększa ryzyko hipokaliemii i może niekorzystnie wpływać na dostarczanie tlenu do tkanek.
• Ciężka hipofosfatemia może prowadzić do rozpadu mięśni szkieletowych, zespołu hemolityczno-mocznicowego, osłabienia i porażenia mięś ni. Podawanie fosforanów wiąże się z ryzykiem hipokalcemii, jednak roztwory dożylne, zawierające mieszaninę fosforanu potasu i innej soli potasu (chlorku lub octanu) w stosunku 50:50 zwykle umożliwiają odpowiednie uzupełnianie fosforanów, równocześnie zapobiegając klinicznie istotnej hipokalcemii.
– Stężenie fosforanów w surowicy należy mierzyć co 3–4 h.
• U chorych z HHS często występuje znaczny niedobór magnezu, brakuje jednak odpowiednich danych wskazujących na korzyści związane z suplementacją tego pierwiastka.
– Uzupełnienie magnezu rozważ u chorych z ciężką hipomagnezemią i hipokalcemią w trakcie leczenia. Zalecana pojedyncza
dawka magnezu wynosi 25–50 mg/kg mc. Należy podać 3–4 takich dawek co 4–6 h z maksymalną prędkością 150 mg/min i 2 g/h.
Powikłania
• Zakładanie cewnika do żył głównych u chorych z HHS często wiąże się z ryzykiem żylnej choroby zakrzepowej.69 U dorosłych sugerowano profilaktyczne podawanie heparyny w małej dawce, ale jak dotąd brakuje danych potwierdzających korzyści z takiego postępowania. Leczenie heparyną należy zarezerwować dla dzieci wymagających założenia dożylnego cewnika centralnego w celu monitorowania parametrów fizjologicznych lub uzyskania dostępu dożylnego oraz unieruchomionych przez ponad 24–48 h.32 Dożylnego cewnika centralnego nie należy używać do podawania wlewu insuliny, ponieważ duża przestrzeń martwa cewnika może prowadzić do błędów w jej dawkowaniu.
• U dzieci z HHS może dojść do rozpadu mięśni szkieletowych, prowadzącego do ostrej niewydolności nerek, ciężkiej hiperkaliemii, hipokalcemii oraz obrzęku mięśni, co z kolei wywołuje zespół ciasnoty przedziałów powięziowych.222 Klasyczna triada objawów rozpadu mięśni szkieletowych obejmuje ból i osłabienie mięśni oraz ciemne zabarwienie moczu. W celu wczesnego wykrycia tego powikłania zaleca się kontrolowanie stężenia kinazy kreatynowej co 2–3 h.
• Z nieznanych powodów u kilkorga dzieci z HHS wystąpiły objawy podobne do objawów hipertermii złośliwej. Powikłanie to charakteryzuje się dużą śmiertelnością.26,223-225 Chorych z gorączką i towarzyszącym zwiększeniem stężenia kinazy kreatynowej można leczyć dantrolenem, który zmniejsza uwalnianie wapnia z siateczki endoplazmatycznej szorstkiej i stabilizuje metabolizm wapnia w komórkach mięśniowych. Mimo to spośród 3 chorych z HHS leczonych dantrolenem przeżył tylko 1.223,225
• Zaburzenia psychiczne są częste u dorosłych z osmolalnością surowicy przekraczającą 330 mOsm/kg, jednak obrzęk mózgu występuje rzadko.28 Spośród 96 chorych z HHS opisanych w piśmiennictwie do 2010 roku, z których 32 zmarło, tylko u 1 rozwinął się obrzęk mózgu (Rosenbloom, informacja uzyskana osobiście). Zaburzenia psychiczne po skutecznym zmniejszeniu hiperosmolalności występują rzadko, a wszystkich chorych z takimi objawami należy poddać szybkiej diagnostyce.
Współwystępowanie HHS i DKA
W leczeniu należy uwzględnić możliwe powikłania zarówno DKA, jak i HHS. Należy ściśle kontrolować stan psychiczny chorego, a żeby leczenie było skuteczne, konieczne są częste oceny układu krążenia i bilansu płynów. Tempo podawania płynów i elektrolitów w celu utrzymania odpowiedniej objętości płynu w łożysku naczyniowym zwykle przekracza wartości stosowane w typowych przypadkach DKA. W celu odwrócenia ketozy i zahamowania glukoneogenezy konieczne jest podanie insuliny, jednak rozpoczęcie jej wlewu należy opóźnić do czasu podania wstępnego bolusu płynów i ustabilizowania układu krążenia. Należy ściśle kontrolować stężenie potasu i fosforanów w surowicy zgodnie z opisanymi powyżej zasadami postępowania w HHS.
Zapobieganie nawrotom DKA
Kompleksowe postępowanie w przypadku DKA obejmuje ustalenie przyczyny tego epizodu i podjęcie próby leczenia przyczynowego.
• W większości przypadków przyczyną DKA jest niezamierzone lub celowe pominięcie dawki insuliny.
• Najczęstszą przyczyną DKA u osób stosujących osobiste pompy insulinowe jest niepodanie dodatkowej dawki insuliny za pomocą wstrzykiwacza lub strzykawki w przypadku wystąpienia hiperglikemii i hiperketonemii lub ketonurii.
• W porównaniu z oceną ketonurii, oznaczenie stężenia BOHB we krwi w warunkach domowych zmniejsza liczbę zgłoszeń do szpitala z powodu cukrzycy (zarówno wizyt na szpitalnych oddziałach ratunkowych, jak i hospitalizacji) dzięki wczesnemu wykryciu i leczeniu ketozy.226 Pomiary stężenia BOHB we krwi mogą mieć szczególną wartość w zapobieganiu DKA u chorych stosujących osobiste pompy insulino- we, ponieważ przerwanie podawania insuliny szybko prowadzi do ketozy.
– Wynik testu na obecność związków ketonowych w moczu (nitroprusydek sodu wykrywa jedynie acetooctan i aceton) i wartość stężenia BOHB w surowicy mogą być rozbieżne. Stężenie BOHB może się bowiem zwiększyć do wartości typowych dla DKA, podczas gdy wynik badania moczu jest ujemny lub wskazuje jedynie na śladową lub niewielką ketonurię.227
• Chorzy na cukrzycę zwykle pomijają dawki insuliny z ważnych przyczyn psychospołecznych, do których należy:
– próba zmniejszenia masy ciała u nastoletnich dziewcząt z zaburzeniami odżywiania
– sposób ucieczki przed trudnymi sytuacjami w domu lub przemocą domową
– depresja lub inne przyczyny niezdolności pacjenta do samodzielnego leczenia cukrzycy.
• W celu rozpoznania psychospołecznych przyczyn wpływających na rozwój DKA należy się skonsultować z pracownikiem środowiskowym/socjalnym zajmującym się zaburzeniami zdrowia psychicznego lub psychologiem klinicznym.
• Zakażenie jest rzadką przyczyną DKA, jeżeli pacjent/rodzina są odpowiednio przeszkoleni w zakresie leczenia cukrzycy, a zespół diabetologiczny zapewnia im właściwą opiekę z możliwością korzystania z całodobowej informacji telefonicznej.228-230
• Pomijaniu dawek insuliny można zapobiegać, wprowadzając kompleksowe programy obejmujące edukację, ocenę psychospołeczną oraz leczenie skojarzone z nadzorowaniem podawania insuliny przez dorosłych.231
– Rodzice i chorzy powinni się uczyć, w jaki sposób rozpoznawać rozwijającą się DKA i leczyć ją za pomocą dodatkowych dawek szybko działającego analogu lub insuliny krótko działającej oraz płynów przyjmowanych doustnie.
– Rodziny powinni mieć dostęp do całodobowej pomocy telefonicznej, zapewniającej porady i pomoc w leczeniu w nagłych przypadkach.228
– Jeżeli insulinę podaje odpowiedzialna osoba dorosła, częstotliwość nawrotów DKA może się zmniejszyć nawet 10-krotnie.231
Konflikt interesów: Autorzy nie zgłosili konfliktu interesów.
b Powikłania często występujące w przebiegu HHS i często prowadzące do zgonu (p. 32. pozycja piśmiennictwa). Patofizjologię i leczenie HHS opisano w dalszej części artykułu.
Piśmiennictwo:
1. Foster D.W., McGarry J.D.: The metabolic derangements and treatment of diabetic ketoacidosis. N. Engl. J. Med., 1983: 309: 159–1692. Kitabchi A.E., Umpierrez G.E., Murphy M.B., Kreisberg R.A.: Hyperglycemic crises in adult patients with diabetes: a consensus statement from the American Diabetes Association. Diabetes Care, 2006; 29: 2739–2748
3. Hanas R., Lindgren F., Lindblad B.: A 2-year national population study of pediatric ke- toacidosis in Sweden: predisposing conditions and insulin pump use. Pediatr. Diabetes, 2009; 10: 33–37
4. Cox K., Cocchi M.N., Salciccioli J.D., Carney E., Howell M., Donnino M.W.: Prevalence and significance of lactic acidosis in diabetic ketoacidosis. J. Crit. Care, 2012; 27: 132–137
5. Deeter K.H., Roberts J.S., Bradford H., et al.: Hypertension despite dehydration during severe pediatric diabetic ketoacidosis. Pediatr. Diabetes, 2011; 12 (4 Pt 1): 295–301
6. McDonnell C.M., Pedreira C.C., Vadamalayan B., Cameron F.J., Werther G.A.: Diabetic ketoacidosis, hyperosmolarity and hypernatremia: are highcarbohydrate drinks worsening initial presentation? Pediatr. Diabetes, 2005; 6: 90–94
7. Carlotti A.P., St George-Hyslop C., Guerguerian A.M., Bohn D., Kamel K.S., Halperin M.: Occult risk factor for the development of cerebral edema in children with diabetic ketoacidosis: possible role for stomach emptying. Pediatr. Diabetes, 2009; 10: 522–533
8. Atchley D., Loeb R., Richards D. Jr, Benedict E., Driscoll M.: On diabetic ketoacidosis: a detailed study of electrolyte balances following the withdrawal and reestablishment of insulin therapy. J. Clin. Invest., 1933; 12: 297–326
9. Nabarro J., Spencer A., Stowers J.: Metabolic studies in severe diabetic ketosis. Q J. Med., 1952; 82: 225–248
10. Butler A., Talbot N., Burnett C., Stanbury J., MacLachlan E.: Metabolic studies in diabetic coma. Trans. Assoc. Am. Physicians, 1947; 60: 102–109
11. Danowski T., Peters J., Rathbun J., Quashnock J., Greenman L.: Studies in diabetic aci- dosis and coma, with particular emphasis on the retention of administered potassium. J. Clin. Invest., 1949; 28: 1–9
12. Darrow D., Pratt E.: Retention of water and electrolyte during recovery in a patient with diabetic acidosis. J. Pediatr., 1952; 41: 688–696
13. Holliday M.A., Segar W.E.: The maintenance need for water in parenteral fluid therapy. Pediatrics, 1957; 19: 823–832
14. Friedman A.L.: Pediatric hydration therapy: historical review and a new approach. Kidney Int., 2005; 67: 380–388
15. Hendricks K., Duggan C. (eds): Manual of Pediatric Nutrition. 4th edn. Hamilton: BC Decker, 2005
16. Darrow D.C.: The physiologic basis for estimating requirements for parenteral fluids. Pediatr. Clin. North Am., 1959; 6: 29–41
17. Dunger D.B., Sperling M.A., Acerini C.L., et al.: ESPE/LWPES consensus statement on diabetic ketoacidosis in children and adolescents. Arch. Dis. Child., 2004; 89: 188–194
18. Sheikh-Ali M., Karon B.S., Basu A., et al.: Can serum beta-hydroxybutyrate be used to diagnose diabetic ketoacidosis? Diabetes Care, 2008; 31: 643–647
19. Burge M.R., Hardy K.J., Schade D.S.: Short-term fasting is a mechanism for the development of euglycemic ketoacidosis during periods of insulin deficiency. J. Clin. Endocrinol. Metab., 1993; 76: 1192–1198
20. Pinkey J.H., Bingley P.J., Sawtell P.A., Dunger D.B., Gale E.A.: Presentation and progress of childhood diabetes mellitus: a prospective population-based study. The Bart’s-Oxford Study Group. Diabetologia, 1994; 37: 70–74
21. Fazeli Farsani S., van der Aa M.P., van der Vorst M.M., Knibbe C.A., de Boer A.: Global trends in the incidence and prevalence of type 2 diabetes in children and adolescents: a systematic review and evaluation of methodological approaches. Diabetologia, 2013; 56: 1471–1488
22. American Diabetes Association: Type 2 diabetes in children and adolescents. (Consensus statement). Diabetes Care, 2000;
23: 381–389 23. Rewers A., Klingensmith G., Davis C., et al.: Presence of diabetic ketoacidosis at diagnosis of diabetes mellitus in youth: the Search for Diabetes in Youth Study. Pediatrics, 2008; 121: e1258–e1266
24. Gungor N., Hannon T., Libman I., Bacha F., Arslanian S.: Type 2 diabetes mellitus in youth: the complete picture to date. Pediatr. Clin. North Am., 2005; 52: 1579–1609
25. Chase H.P., Garg S.K., Jelley D.H.: Diabetic ketoacidosis in children and the role of outpatient management. Pediatr. Rev., 1990; 11: 297–304
26. Morales A.E., Rosenbloom A.L.: Death caused by hyperglycemic hyperosmolar state at the onset of type 2 diabetes. J. Pediatr., 2004; 144: 270–273
27. Canarie M.F., Bogue C.W., Banasiak K.J., Weinzimer S.A., Tamborlane W.V.: Decompensated hyperglycemic hyperosmolarity without significant ketoacidosis in the adolescent and young adult population. J. Pediatr. Endocrinol. Metab., 2007; 20: 1115–1124
28. Rosenbloom A.L.: Hyperglycemic hyperosmolar state: an emerging pediatric problem. J. Pediatr., 2010; 156: 180–184
29. Bagdure D., Rewers A., Campagna E., Sills M.R.: Epidemiology of hyperglycemic hyperosmolar syndrome in children hospitalized in USA. Pediatr. Diabetes, 2013; 14: 18–24
30. Temple I.K., Shield J.P.: 6q24 transient neonatal diabetes. Rev. Endocr. Metab. Disord., 2010; 11: 199–204
31. Kitabchi A.E., Nyenwe E.A.: Hyperglycemic crises in diabetes mellitus: diabetic ketoacidosis and hyperglycemic hyperosmolar state. Endocrinol. Metab. Clin. North Am., 2006; 35: 725–751
32. Zeitler P., Haqq A., Rosenbloom A., Glaser N.: Hyperglycemic hyperosmolar syndrome in children: pathophysiological considerations and suggested guidelines for treatment. J. Pediatr., 2011; 158: 9–14
33. Levy-Marchal C., Papoz L., de Beaufort C., et al.: Clinical and laboratory features of type 1 diabetic children at the time of diagnosis. Diabet. Med., 1992; 9: 279–284
34. Komulainen J., Lounamaa R., Knip M., Kaprio E.A., Akerblom H.K.: Ketoacidosis at the di- agnosis of type 1 (insulin dependent) diabetes mellitus is related to poor residual beta cell function. Childhood Diabetes in Finland Study Group. Arch. Dis. Child., 1996; 75: 410–415
35. Levy-Marchal C., Patterson C.C., Green A.: Geographical variation of presentation at diagnosis of type I diabetes in children: the EURODIAB study. European and Dibetes. Diabetologia, 2001; 44 (Suppl. 3): B75–B80
36. Hanas R., Lindgren F., Lindblad B.: Diabetic ketoacidosis and cerebral edema in Sweden – a 2-year population study. Diabet. Med., 2007; 24: 1080–1085
37. Rodacki M., Pereira J.R., Nabuco de Oliveira A.M., et al.: Ethnicity and young age influence the frequency of diabetic ketoacidosis at the onset of type 1 diabetes. Diabetes Res. Clin. Pract., 2007; 78: 259–262
38. Usher-Smith J.A., Thompson M., Ercole A., Walter F.M.: Variation between countries in the frequency of diabetic ketoacidosis at first presentation of type 1 diabetes in children: a systematic review. Diabetologia, 2012; 55: 2878–2894
39. Quinn M., Fleischman A., Rosner B., Nigrin D.J., Wolfsdorf J.I.: Characteristics at diagnosis of type 1 diabetes in children younger than 6 years. J. Pediatr., 2006; 148: 366–371
40. Bui H., To T., Stein R., Fung K., Daneman D.: Is diabetic ketoacidosis at disease onset a result of missed diagnosis? J. Pediatr., 2010; 156: 472–477
41. Szypowska A., Skorka A.: The risk factors of ketoacidosis in children with newly diagnosed type 1 diabetes mellitus. Pediatr. Diabetes, 2011; 12 (4 Pt 1): 302–306
42. Komulainen J., Kulmala P., Savola K., et al.: Clinical, autoimmune, and genetic character- istics of very young children with type 1 diabetes. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes Care, 1999; 22: 1950–1955
43. Usher-Smith J.A., Thompson M.J., Sharp S.J., Walter F.M.: Factors associated with the presence of diabetic ketoacidosis at diagnosis of diabetes in children and young adults: a systematic review. BMJ, 2011; 343: d4092
44. Rosilio M., Cotton J.B., Wieliczko M.C., et al.: Factors associated with glycemic control. A cross-sectional nationwide study in 2,579 French children with type 1 diabetes. The French Pediatric Diabetes Group [see comments]. Diabetes Care, 1998; 21: 1146–1153
45. Smith C.P., Firth D., Bennett S., Howard C., Chisholm P.: Ketoacidosis occurring in newly diagnosed and established diabetic children. Acta Paediatr., 1998; 87: 537–541
46. Morris A.D., Boyle D.I., McMahon A.D., Greene S.A., MacDonald T.M., Newton R.W.: Adherence to insulin treatment, glycaemic control, and ketoacidosis in insulin-dependent diabetes mellitus. The DARTS/MEMO Collaboration. Diabetes Audit and Research in Tayside Scotland. Medicines Monitoring Unit. Lancet, 1997; 350: 1505–1510
47. Rewers A., Chase H.P., Mackenzie T., et al.: Predictors of acute complications in children with type 1 diabetes. JAMA, 2002; 287: 2511–2518
48. Cengiz E., Xing D., Wong J.C., et al.: Severe hypoglycemia and diabetic ketoacidosis among youth with type 1 diabetes in the T1D Exchange clinic registry. Pediatr. Diabetes, 2013; 14: 447–454
49. Cope J.U., Morrison A.E., Samuels-Reid J.: Adolescent use of insulin and patient-controlled analgesia pump technology: a 10-year Food and Drug Administration retrospective study of adverse events. Pediatrics, 2008; 121: e1133–e1138
50. Kleinman M.E., Chameides L., Schexnayder S.M., et al.: Pediatric advanced life support: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Pediatrics, 2010; 126: e1361–e1399
51. Kleinman M.E., de Caen A.R., Chameides L., et al.: Pediatric basic and advanced life support: 2010 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science with Treatment Recommendations. Pediatrics, 2010; 126: e1261–e1318
52. Wiggam M.I., O’Kane M.J., Harper R., et al.: Treatment of diabetic ketoacidosis using normalization of blood 3-hydroxybutyrate concentration as the endpoint of emergency management. A randomized controlled study. Diabetes Care, 1997; 20: 1347–1352
53. Vanelli M., Chiari G., Capuano C., Iovane B., Bernardini A., Giacalone T.: The direct measurement of 3-beta-hydroxy butyrate enhances the management of diabetic ketoacidosis in children and reduces time and costs of treatment. Diabetes Nutr. Metab., 2003; 16 (5–6): 312–316
54. Ham M.R., Okada P., White P.C.: Bedside ketone determination in diabetic children with hyperglycemia and ketosis in the acute care setting. Pediatr. Diabetes, 2004; 5: 39–43
55. Rewers A., McFann K., Chase H.P.: Bedside monitoring of blood beta-hydroxybutyrate levels in the management of diabetic ketoacidosis in children. Diabetes Technol. Ther., 2006; 8: 671–676
56. Prisco F., Picardi A., Iafusco D., et al.: Blood ketone bodies in patients with recent-onset type 1 diabetes (a multicenter study). Pediatr. Diabetes, 2006; 7: 223–228
57. Noyes K.J., Crofton P., Bath L.E., et al.: Hydroxybutyrate near-patient testing to evaluate a new end-point for intravenous insulin therapy in the treatment of diabetic ketoacidosis in children. Pediatr. Diabetes, 2007; 8: 150–156
58. Klocker A.A., Phelan H., Twigg S.M., Craig M.E.: Blood beta-hydroxybutyrate vs. urine acetoacetate testing for the prevention and management of ketoacidosis in Type 1 diabetes: a systematic review. Diabet. Med., 2013; 30: 818–824
59. Koves I.H., Neutze J., Donath S., et al.: The accuracy of clinical assessment of dehydra- tion during diabetic ketoacidosis in childhood. Diabetes Care, 2004; 27: 2485–2487
60. Sottosanti M., Morrison G.C., Singh R.N., et al.: Dehydration in children with diabetic ketoacidosis: a prospective study. Arch. Dis. Child., 2012; 97: 96–100
61. Ugale J., Mata A., Meert K.L., Sarnaik A.P.: Measured degree of dehydration in children and adolescents with type 1 diabetic ketoacidosis. Pediatr. Crit. Care Med., 2012; 13: e103–e107
62. Steiner M.J., DeWalt D.A., Byerley J.S.: Is this child dehydrated? JAMA, 2004; 291: 2746–2754
63. Teasdale G., Jennett B.: Assessment of coma and impaired consciousness. A practical scale. Lancet, 1974; 2: 81–84
64. Flood R.G., Chiang V.W.: Rate and prediction of infection in children with diabetic ketoacidosis. Am. J. Emerg. Med., 2001; 19: 270–273
65. Malone J.I., Brodsky S.J.: The value of electrocardiogram monitoring in diabetic keto- acidosis. Diabetes Care, 1980; 3: 543–547
66. Soler N.G., Bennett M.A., Fitzgerald M.G., Malins J.M.: Electrocardiogram as a guide topotassium replacement in diabetic ketoacidosis. Diabetes, 1974; 23: 610–615
67. Tasker R.C., Lutman D., Peters M.J.: Hyperventilation in severe diabetic ketoacidosis. Pediatr. Crit. Care Med., 2005; 6: 405–411
68. Davids M.R., Edoute Y., Stock S., Halperin M.L.: Severe degree of hyperglycaemia: insights from integrative physiology. QJM, 2002; 95: 113–124
69. Gutierrez J.A., Bagatell R., Samson M.P., Theodorou A.A., Berg R.A.: Femoral central venous catheterassociated deep venous thrombosis in children with diabetic ketoacidosis. Crit. Care Med., 2003; 31: 80–83
70. Worly J.M., Fortenberry J.D., Hansen I., Chambliss C.R., Stockwell J.: Deep venous thrombosis in children with diabetic ketoacidosis and femoral central venous catheters. Pediatrics, 2004; 113 (1 Pt 1): e57–e60
71. Monroe K.W., King W., Atchison J.A.: Use of PRISM scores in triage of pediatric patients with diabetic ketoacidosis. Am. J. Manag. Care, 1997; 3: 253–258
72. Bonadio W.A., Gutzeit M.F., Losek J.D., Smith D.S.: Outpatient management of diabetic ketoacidosis. Am. J. Dis. Child., 1988; 142: 448–450
73. Linares M.Y., Schunk J.E., Lindsay R.: Laboratory presentation in diabetic ketoacidosis and duration of therapy. Pediatr. Emerg. Care, 1996; 12: 347–351
74. Yu H.Y., Agus M., Kellogg M.D.: Clinical utility of Abbott Precision Xceed Pro(R) ketone meter in diabetic patients. Pediatr. Diabetes, 2011; 12: 649–655
75. Lutfi R., Huang J., Wong H.R.: Plasmapheresis to treat hypertriglyceridemia in a child with diabetic ketoacidosis and pancreatitis. Pediatrics, 2012; 129: e195–e198
76. Halperin M.L., Goldstein M.B.: Fluid, electrolyte, and acid-base physiology. 3rd edn. Philadelphia: Saunders, 1999
77. Love A.H., Phillips R.A.: Measurement of dehydration in cholera. J. Infect. Dis., 1969; 119: 39–42
78. Harris G.D., Fiordalisi I.: Physiologic management of diabetic ketoacidemia. A 5-year prospective pediatric experience in 231 episodes. Arch. Pediatr. Adolesc. Med., 1994; 148: 1046–1052
79. Napolova O., Urbach S., Davids M.R., Halperin M.L.: Assessing the degree of extracellular fluid volume contraction in a patient with a severe degree of hyperglycaemia. Nephrol. Dial. Transplant., 2003; 18: 2674–2677
80. Halperin M.L., Maccari C., Kamel K.S., Carlotti A.P., Bohn D.: Strategies to diminish the danger of cerebral edema in a pediatric patient presenting with diabetic ketoacidosis. Pediatr. Diabetes, 2006; 7: 191–195
81. Katz M.A.: Hyperglycemia-induced hyponatremia – calculation of expected serum sodium depression. N. Engl. J. Med., 1973; 289: 843–844
82. Hillier T.A., Abbott R.D., Barrett E.J.: Hyponatremia: evaluating the correction factor for hyperglycemia. Am. J. Med., 1999; 106: 399–403
83. Harris G.D., Fiordalisi I., Harris W.L., Mosovich L.L., Finberg L.: Minimizing the risk of brain herniation during treatment of diabetic ketoacidemia: a retrospective and prospective study. J. Pediatr., 1990; 117: 22–31
84. Hale P.M., Rezvani I., Braunstein A.W., Lipman T.H., Martinez N., Garibaldi L.: Factors predicting cerebral edema in young children with diabetic ketoacidosis and new onset type I diabetes. Acta Paediatr., 1997; 86: 626–631
85. Glaser N., Barnett P., McCaslin I., et al.: Risk factors for cerebral edema in children with diabetic ketoacidosis. The Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. N. Engl. J. Med., 2001; 344: 264–269
86. Brown T.B.: Cerebral oedema in childhood diabetic ketoacidosis: is treatment a factor? Emerg. Med. J., 2004; 21: 141–144
87. Glaser N.S., Ghetti S., Casper T.C., Dean J.M., Kuppermann N.: Pediatric diabetic ketoacidosis, fluid therapy, and cerebral injury: the design of a factorial randomized controlled trial. Pediatr. Diabetes, 2013; 14: 435–446
88. Edge J.A., Jakes R.W., Roy Y., et al.: The UK case-control study of cerebral oedema complicating diabetic ketoacidosis in children. Diabetologia, 2006; 49: 2002–2009
89. Dunger D.B., Sperling M.A., Acerini C.L., et al.: European Society for Paediatric Endocrinology/Lawson Wilkins Pediatric Endocrine Society consensus statement on diabetic ketoacidosis in children and adolescents. Pediatrics, 2004; 113: e133–e140
90. Adrogue H.J., Barrero J., Eknoyan G.: Salutary effects of modest fluid replacement in the treatment of adults with diabetic ketoacidosis. Use in patients without extreme volume deficit. JAMA, 1989; 262: 2108–2113
91. Mel J.M., Werther G.A.: Incidence and outcome of diabetic cerebral oedema in childhood: are there predictors? J. Paediatr. Child Health, 1995; 31: 17–20
92. Wagner A., Risse A., Brill H.L., et al.: Therapy of severe diabetic ketoacidosis. Zero-mortality under very-lowdose insulin application. Diabetes Care, 1999; 22: 674–677
93. Toledo J.D., Modesto V., Peinador M., et al.: Sodium concentration in rehydration fluids for children with ketoacidotic diabetes: effect on serum sodium concentration. J. Pediatr., 2009; 154: 895–900
94. Rother K.I., Schwenk W.F. 2nd: Effect of rehydration fluid with 75 mmol/L of sodium on serum sodium concentration and serum osmolality in young patients with diabetic ketoacidosis. Mayo Clin. Proc., 1994; 69: 1149–1153
95. Felner E.I., White P.C.: Improving management of diabetic ketoacidosis in children. Pediatrics, 2001; 108: 735–740
96. Basnet S., Venepalli P.K., Andoh J., Verhulst S., Koirala J.: Effect of normal saline and half normal saline on serum electrolytes during recovery phase of diabetic ketoacidosis. J. Intensive Care Med., 2014; 29: 38–42
97. Fiordalisi I., Novotny W.E., Holbert D., Finberg L., Harris G.D.: An 18-year prospective study of pediatric diabetic ketoacidosis: an approach to minimizing the risk of brain herniation during treatment. Pediatr. Diabetes, 2007; 8: 142–149
98. White P.C., Dickson B.A.: Low morbidity and mortality in children with diabetic ketoacidosis treated with isotonic fluids. J. Pediatr., 2013; 163: 761–766
99. Hoorn E.J., Carlotti A.P., Costa L.A., et al.: Preventing a drop in effective plasma osmolality to minimize the likelihood of cerebral edema during treatment of childrenwith diabetic ketoacidosis. J. Pediatr., 2007; 150: 467–473
100. Duck S.C., Wyatt D.T.: Factors associated with brain herniation in the treatment of diabetic ketoacidosis. J. Pediatr., 1988; 113: 10–14
101. Adrogue H.J., Wilson H., Boyd A.E. 3rd, Suki W.N., Eknoyan G.: Plasma acid–base patterns in diabetic ketoacidosis. N. Engl. J. Med., 1982; 307: 1603–1610
102. Adrogue H.J., Eknoyan G., Suki W.K.: Diabetic ketoacidosis: role of the kidney in the acidbase homeostasis re-evaluated. Kidney Int., 1984; 25: 591–598
103. Taylor D., Durward A., Tibby S.M., et al.: The influence of hyperchloraemia on acid base interpretation in diabetic ketoacidosis. Intensive Care Med., 2006; 32: 295–301
104. Durward A., Skellett S., Mayer A., Taylor D., Tibby S.M., Murdoch I.A.: The value of the chloride: sodium ratio in differentiating the aetiology of metabolic acidosis. Intensive Care Med., 2001; 27: 828–835
105. Oh M.S., Carroll H.J., Goldstein D.A., Fein I.A.: Hyperchloremic acidosis during the recovery phase of diabetic ketosis. Ann. Intern. Med., 1978; 89: 925–927
106. Oh M.S., Banerji M.A., Carroll H.J.: The mechanism of hyperchloremic acidosis during the recovery phase of diabetic ketoacidosis. Diabetes, 1981; 30: 310–313
107. Oh M.S., Carroll H.J., Uribarri J.: Mechanism of normochloremic and hyperchloremic acidosis in diabetic ketoacidosis. Nephron, 1990; 54: 1–6
108. Chua H.R., Venkatesh B., Stachowski E., et al.: Plasma-Lyte 148 vs 0,9% saline for fluid resuscitation in diabetic ketoacidosis. J. Crit. Care, 2012; 27: 138–145
109. Waldhausl W., Kleinberger G., Korn A., Dudczak R., Bratusch-Marrain P., Nowotny P.: Severe hyperglycemia: effects of rehydration on endocrine derangements and blood glucose concentration. Diabetes, 1979; 28: 577–584
110. Owen O.E., Licht J.H., Sapir D.G.: Renal function and effects of partial rehydration during diabetic ketoacidosis. Diabetes, 1981; 30: 510–518
111. Luzi L., Barrett E.J., Groop L.C., Ferrannini E., DeFronzo R.A.: Metabolic effects of low-dose insulin therapy on glucose metabolism in diabetic ketoacidosis. Diabetes, 1988; 37: 1470–1477
112. Kitabchi A.E.: Low-dose insulin therapy in diabetic ketoacidosis: fact or fiction? Diabetes Metab. Rev., 1989; 5: 337–363
113. Martin M.M., Martin A.A.: Continuous low-dose infusion of insulin in the treatment of diabetic ketoacidosis in children. J. Pediatr., 1976; 89: 560–564
114. Edwards G.A., Kohaut E.C., Wehring B., Hill L.L.: Effectiveness of low-dose continuous intravenous insulin infusion in diabetic ketoacidosis. A prospective comparative study. J. Pediatr., 1977; 91: 701–705
115. Drop S.L., Duval-Arnould J.M., Gober A.E., Hersh J.H., McEnery P.T., Knowles H.C.: Lowdose intravenous insulin infusion versus subcutaneous insulin injection: a controlled comparative study of diabetic ketoacidosis. Pediatrics, 1977; 59: 733–738
116. Lightner E.S., Kappy M.S., Revsin B.: Low-dose intravenous insulin infusion in patients with diabetic ketoacidosis: biochemical effects in children. Pediatrics, 1977; 60: 681–688
117. Perkin R.M., Marks J.F.: Low-dose continuous intravenous insulin infusion in childhood diabetic ketoacidosis. Clin. Pediatr. (Phila), 1979; 18: 540, 545–540, 548
118. Kappy M.S., Lightner E.S.: Low-dose intravenous insulin in the treatment of diabetic ketoacidosis. Am. J. Dis. Child., 1979; 133: 523–525
119. Burghen G.A., Etteldorf J.N., Fisher J.N., Kitabchi A.Q.: Comparison of high-dose and low-dose insulin by continuous intravenous infusion in the treatment of diabetic ketoacidosis in children. Diabetes Care, 1980; 3: 15–20
120. Kitabchi A.E., Umpierrez G.E., Fisher J.N., Murphy M.B., Stentz F.B.: Thirty years of personal experience in hyperglycemic crises: diabetic ketoacidosis and hyperglycemic hyperosmolar state. J. Clin. Endocrinol. Metab., 2008; 93: 1541–1552
121. Lindsay R., Bolte R.G.: The use of an insulin bolus in low-dose insulin infusion for pediatric diabetic ketoacidosis. Pediatr. Emerg. Care, 1989; 5: 77–79
122. Van der Meulen J.A., Klip A., Grinstein S.: Possible mechanism for cerebral oedema in diabetic ketoacidosis. Lancet, 1987; 2: 306–308
123. Soler N.G., FitzGerald M.G., Wright A.D., Malins J.M.: Comparative study of different insulin regimens in management of diabetic ketoacidosis. Lancet, 1975; 2: 1221–1224
124. Puttha R., Cooke D., Subbarayan A., et al.: Low dose (0,05 units/kg/h) is comparable with standard dose (0,1 units/kg/h) intravenous insulin infusion for the initial treatment of diabetic ketoacidosis in children with type 1 diabetes-an observational study. Pediatr. Diabetes, 2010; 11: 12–17
125. Al Hanshi S., Shann F.: Insulin infused at 0,05 versus 0,1 units/kg/hr in children admitted to intensive care with diabetic ketoacidosis. Pediatr. Crit. Care Med., 2011; 12: 137–140
126. Wang J., Barbry P., Maiyar A.C., et al.: SGK integrates insulin and mineralocorticoid regulation of epithelial sodium transport. Am. J. Physiol. Renal. Physiol., 2001; 280: F303–F313
127. Tallini N.Y., Stoner L.C.: Amiloride-sensitive sodium current in everted Ambystoma initial collecting tubule: short-term insulin effects. Am. J. Physiol. Cell Physiol., 2002; 283: C1171–C1181
128. McCormick J.A., Bhalla V., Pao A.C., Pearce D.: SGK1: a rapid aldosterone-induced regulator of renal sodium reabsorption. Physiology, 2005; 20: 134–139
129. Tiwari S., Nordquist L., Halagappa V.K., Ecelbarger C.A.: Trafficking of ENaC subunits in response to acute insulin in mouse kidney. Am. J. Physiol. Renal. Physiol., 2007; 293: F178–F185
130. Frindt G., Palmer L.G.: Effects of insulin on Na and K transporters in the rat CCD. Am. J. Physiol. Renal. Physiol., 2012; 302: F1227–F1233
131. Carlotti A.P., St George-Hyslop C., Bohn D., Halperin M.L.: Hypokalemia during treat- ment of diabetic ketoacidosis: clinical evidence for an aldosterone-like action of insulin. J. Pediatr., 2013; 163: 207–212 e201
132. Fisher J.N., Shahshahani M.N., Kitabchi A.E.: Diabetic ketoacidosis: low-dose insulin therapy by various routes. N. Engl. J. Med., 1977; 297: 238–241
133. Sacks H.S., Shahshahani M., Kitabchi A.E., Fisher J.N., Young R.T.: Similar responsiveness of diabetic ketoacidosis to low-dose insulin by intramuscular injection and albumin-free infusion. Ann. Intern. Med., 1979; 90: 36–42
134. Umpierrez G.E., Latif K., Stoever J., et al.: Efficacy of subcutaneous insulin lispro versus continuous intravenous regular insulin for the treatment of patients with diabetic ketoacidosis. Am. J. Med., 2004; 117: 291–296
135. Umpierrez G.E., Cuervo R., Karabell A., Latif K., Freire A.X., Kitabchi A.E.: Treatment of di- abetic ketoacidosis with subcutaneous insulin aspart. Diabetes Care, 2004; 27: 1873–1878
136. Della Manna T., Steinmetz L., Campos P.R., et al.: Subcutaneous use of a fast-acting insulin analog: an alternative treatment for pediatric patients with diabetic ketoacidosis. Diabetes Care, 2005; 28: 1856–1861
137. Adrogue H.J., Lederer E.D., Suki W.N., Eknoyan G.: Determinants of plasma potassium levels in diabetic ketoacidosis. Medicine (Baltimore), 1986; 65: 163–172
138. DeFronzo R.A., Felig P., Ferrannini E., Wahren J.: Effect of graded doses of insulin on splanchnic and peripheral potassium metabolism inman. Am. J. Physiol., 1980; 238: E421–E427
139. Tattersall R.B.: A paper which changed clinical practice (slowly). Jacob Holler on potassium deficiency in diabetic acidosis (1946). Diabet. Med., 1999; 16: 978–984
140. Nabarro J.D., Spencer A.G., Stowers J.M.: Treatment of diabetic ketosis. Lancet, 1952; 1: 983–989 141. Guest G.: Organic phosphates of the blood andmineral metabolism in diabetic acidosis. Am. J. Dis. Child., 1942; 64: 401–412
142. Guest G., Rapoport S.: Electrolytes of blood plasma and cells in diabetic acidosis and during recovery. Proc. Am. Diabetes Assoc., 1947; 7: 95–115
143. Riley M.S., Schade D.S., Eaton R.P.: Effects of insulin infusion on plasma phosphate in diabetic patients. Metabolism, 1979; 28: 191–194
144. Alberti K.G., Emerson P.M., Darley J.H., Hockaday T.D.: 2,3-Diphosphoglycerate and tissue oxygenation in uncontrolled diabetes mellitus. Lancet, 1972; 2: 391–395
145. Knochel J.P.: The pathophysiology and clinical characteristics of severe hypophosphatemia. Arch. Intern. Med., 1977; 137: 203–220
146. O’Connor L.R., Wheeler W.S., Bethune J.E.: Effect of hypophosphatemia on myocardial performance in man. N. Engl. J. Med., 1977; 297: 901–903
147. Gibby O.M., Veale K.E., Hayes T.M., Jones J.G., Wardrop C.A.: Oxygen availability from the blood and the effect of phosphate replacement on erythrocyte 2,3-diphosphoglycerate and haemoglobin-oxygen affinity in diabetic ketoacidosis. Diabetologia, 1978; 15: 381–385
148. Keller U., Berger W.: Prevention of hypophosphatemia by phosphate infusion during treatment of diabetic ketoacidosis and hyperosmolar coma. Diabetes, 1980; 29: 87–95
149. Wilson H.K., Keuer S.P., Lea A.S., Boyd A.E. 3rd, Eknoyan G.: Phosphate therapy in diabetic ketoacidosis. Arch. Intern. Med., 1982; 142: 517–520
150. Becker D.J., Brown D.R., Steranka B.H., Drash A.L.: Phosphate replacement during treatment of diabetic ketosis. Effects on calcium and phosphorus homeostasis. Am. J. Dis. Child., 1983; 137: 241–246
151. Fisher J.N., Kitabchi A.E.: A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. J. Clin. Endocrinol. Metab., 1983; 57: 177–180
152. Clerbaux T., Reynaert M., Willems E., Frans A.: Effect of phosphate on oxygen-hemoglobin affinity, diphosphoglycerate and blood gases during recovery from diabetic ketoacidosis. Intensive Care Med., 1989; 15: 495–498
153. Weisinger J.R., Bellorin-Font E.: Magnesium and phosphorus. Lancet, 1998; 352: 391–396
154. Singhal P.C., Kumar A., Desroches L., Gibbons N., Mattana J.: Prevalence and predictors of rhabdomyolysis in patients with hypophosphatemia. Am. J. Med., 1992; 92: 458–464
155. Kutlu A.O., Kara C., Cetinkaya S.: Rhabdomyolysis without detectable myoglobulinuria due to severe hypophosphatemia in diabetic ketoacidosis. Pediatr. Emerg. Care, 2011; 27: 537–538
156. Bohannon N.J.: Large phosphate shifts with treatment for hyperglycemia. Arch. Intern. Med., 1989; 149: 1423–1425
157. de Oliveira Iglesias S.B., Pons Leite H., de Carvalho W.B.: Hypophosphatemia-induced seizure in a child with diabetic ketoacidosis. Pediatr. Emerg. Care, 2009; 25: 859–861
158. Zipf W.B., Bacon G.E., Spencer M.L., Kelch R.P., Hopwood N.J., Hawker C.D.: Hypocalcemia, hypomagnesemia, and transient hypoparathyroidism during therapy with potassium phosphate in diabetic ketoacidosis. Diabetes Care, 1979; 2: 265–268
159. Winter R.J., Harris C.J., Phillips L.S., Green O.C.: Diabetic ketoacidosis. Induction of hy- pocalcemia and hypomagnesemia by phosphate therapy. Am. J. Med., 1979; 67: 897–900
160. Hale P.J., Crase J., Nattrass M.: Metabolic effects of bicarbonate in the treatment of diabetic ketoacidosis. Br. Med. J. (Clin. Res. Ed), 1984; 289: 1035–1038
161. Morris L.R., Murphy M.B., Kitabchi A.E.: Bicarbonate therapy in severe diabetic ketoac- idosis. Ann. Intern. Med., 1986; 105: 836–840
162. Okuda Y., Adrogue H.J., Field J.B., Nohara H., Yamashita K.: Counterproductive effects of sodium bicarbonate in diabetic ketoacidosis. J. Clin. Endocrinol. Metab., 1996; 81: 314–320
163. Green S.M., Rothrock S.G., Ho J.D., et al.: Failure of adjunctive bicarbonate to improve outcome in severe pediatric diabetic ketoacidosis. Ann. Emerg. Med., 1998; 31: 41–48
164. Assal J.P., Aoki T.T., Manzano F.M., Kozak G.P.: Metabolic effects of sodium bicarbonate in management of diabetic ketoacidosis. Diabetes, 1974; 23: 405–411
165. Ohman J.L. Jr, Marliss E.B., Aoki T.T., Munichoodappa C.S., Khanna V.V., Kozak G.P.: The cerebrospinal fluid in diabetic ketoacidosis. N. Engl. J. Med., 1971; 284: 283–290
166. Soler N.G., Bennett M.A., Dixon K., FitzGerald M.G., Malins J.M.: Potassium balance during treatment of diabetic ketoacidosis with special reference to the use of bicarbonate. Lancet, 1972; 2: 665–667
167. Lever E., Jaspan J.B.: Sodium bicarbonate therapy in severe diabetic ketoacidosis. Am. J. Med., 1983; 75: 263–268
168. Narins R.G., Cohen J.J.: Bicarbonate therapy for organic acidosis: the case for its continued use. Ann. Intern. Med., 1987; 106: 615–618
169. Curtis J.R., To T., Muirhead S., Cummings E., Daneman D.: Recent trends in hospitaliza- tion for diabetic ketoacidosis in Ontario children. Diabetes Care, 2002; 25: 1591–1596
170. Edge J.A., Ford-Adams M.E., Dunger D.B.: Causes of death in children with insulin dependent diabetes 1990–96. Arch. Dis. Child., 1999; 81: 318–323
171. Decourcey D.D., Steil G.M., Wypij D., Agus M.S.: Increasing use of hypertonic saline overmannitol in the treatment of symptomatic cerebral edema in pediatric diabetic ketoacidosis: an 11-year retrospective analysis of mortality*. Pediatr. Crit. Care Med., 2013; 14: 694–700
172. Daneman D.: Diabetes-related mortality. A pediatrician’s view. Diabetes Care, 2001; 24: 801–802
173. Edge J.A., Hawkins M.M., Winter D.L., Dunger D.B.: The risk and outcome of cerebral oedema developing during diabetic ketoacidosis. Arch. Dis. Child., 2001; 85: 16–22
174. Lawrence S.E., Cummings E.A., Gaboury I., Daneman D.: Population-based study of incidence and risk factors for cerebral edema in pediatric diabetic ketoacidosis. J. Pediatr., 2005; 146: 688–692
175. Ghetti S., Lee J.K., Sims C.E., Demaster D.M., Glaser N.S.: Diabetic ketoacidosis and memory dysfunction in children with type 1 diabetes. J. Pediatr., 2010; 156: 109–114
176. Cooper M.R., Turner R.A. Jr, Hutaff L., Prichard R.: Diabetic keto-acidosis complicated by disseminated intravascular coagulation. South Med. J., 1973; 66: 653–657
177. Keane S., Gallagher A., Ackroyd S., McShane M.A., Edge J.A.: Cerebral venous thrombosis during diabetic ketoacidosis. Arch. Dis. Child., 2002; 86: 204–205
178. Ho J., Mah J.K., Hill M.D., Pacaud D.: Pediatric stroke associated with new onset type 1 diabetesmellitus: case reports and review of the literature. Pediatr. Diabetes, 2006; 7: 116–121
179. Moye J., Rosenbloom A.L., Silverstein J.: Clinical predictors of mucormycosis in children with type 1 diabetes mellitus. J. Pediatr. Endocrinol. Metab., 2002; 15: 1001–1004
180. Watson J.P., Barnett A.H.: Pneumomediastinum in diabetic ketoacidosis. Diabet. Med., 1989; 6: 173–174
181. Haddad N.G., Croffie J.M., Eugster E.A.: Pancreatic enzyme elevations in children with diabetic ketoacidosis. J. Pediatr., 2004; 145: 122–124
182. Glaser N.S., Wootton-Gorges S.L., Buonocore M.H., et al.: Frequency of sub-clinical cerebral edema in children with diabetic ketoacidosis. Pediatr. Diabetes, 2006; 7: 75–80
183. Glaser N.S., Marcin J.P., Wootton-Gorges S.L., et al.: Correlation of clinical and biochemical findings with diabetic ketoacidosis-related cerebral edema in children using magnetic resonance diffusion-weighted imaging. J. Pediatr., 2008; 153: 541–546
184. Krane E.J., Rockoff M.A., Wallman J.K., Wolfsdorf J.I.: Subclinical brain swelling in children during treatment of diabetic ketoacidosis. N. Engl. J. Med., 1985; 312: 1147–1151
185. Hoffman W.H., Steinhart C.M., el Gammal T., Steele S., Cuadrado A.R., Morse P.K.: Cranial CT in children and adolescents with diabetic ketoacidosis. AJNR Am. J. Neuroradiol., 1988; 9: 733–739
186. Sperling M.A.: Cerebral edema in diabetic ketoacidosis: an underestimated complication? Pediatr. Diabetes, 2006; 7: 73–74
187. Arieff A.I., Kleeman C.R.: Studies on mechanisms of cerebral edema in diabetic comas. Effects of hyperglycemia and rapid lowering of plasma glucose in normal rabbits. J. Clin. Invest., 1973; 52: 571–583
188. Arieff A.I., Kleeman C.R.: Cerebral edema in diabetic comas. II. Effects of hyperosmo- lality, hyperglycemia and insulin in diabetic rabbits. J. Clin. Endocrinol. Metab., 1974; 38: 1057–1067
189. Prockop L.D.: Hyperglycemia, polyol accumulation, and increased intracranial pressure. Arch. Neurol., 1971; 25: 126–140
190. Harris G.D., Fiordalisi I., Finberg L.: Safe management of diabetic ketoacidemia. J. Pediatr., 1988; 113: 65–67
191. Glaser N.S., Wootton-Gorges S.L., Marcin J.P., et al.: Mechanism of cerebral edema in children with diabetic ketoacidosis. J. Pediatr., 2004; 145: 164–171
192. Glaser N., Yuen N., Anderson S.E., Tancredi D.J., O’Donnell M.E.: Cerebral metabolic alterations in rats with diabetic ketoacidosis: effects of treatment with insulin and intravenous fluids and effects of bumetanide. Diabetes, 2010; 59: 702–709
193. Yuen N., Anderson S.E., Glaser N., Tancredi D.J., O’Donnell M.E.: Cerebral blood flow and cerebral edema in rats with diabetic ketoacidosis. Diabetes, 2008; 57: 2588–2594
194. Rosenbloom A.L., Schatz D.A., Krischer J.P., et al.: Therapeutic controversy: prevention and treatment of diabetes in children. J. Clin. Endocrinol. Metab., 2000; 85: 494–522
195. Glaser N.: Cerebral injury and cerebral edema in children with diabetic ketoacidosis: could cerebral ischemia and reperfusion injury be involved? Pediatr. Diabetes, 2009; 10: 534–541
196. Hoffman W.H., Stamatovic S.M., Andjelkovic A.V.: Inflammatory mediators and blood brain barrier disruption in fatal brain edema of diabetic ketoacidosis. Brain Res., 2009; 1254: 138–148
197. Vavilala M.S., Richards T.L., Roberts J.S., et al.: Change in blood–brain barrier permeability during pediatric diabetic ketoacidosis treatment*. Pediatr. Crit. Care Med., 2010; 11: 332–338
198. Rosenbloom A.L.: Intracerebral crises during treatment of diabetic ketoacidosis. Diabetes Care, 1990; 13: 22–33
199. Bello F.A., Sotos J.F.: Cerebral oedema in diabetic ketoacidosis in children [letter]. Lancet, 1990; 336: 64
200. Mahoney C.P., Vlcek B.W., Del Aguila M.: Risk factors for developing brain herniation during diabetic ketoacidosis. Pediatr. Neurol., 1999; 21: 721–727
201. Durr J.A., Hoffman W.H., Sklar A.H., el Gammal T., Steinhart C.M.: Correlates of brain edema in uncontrolled IDDM. Diabetes, 1992; 41: 627–632
202. Durward A., Ferguson L.P., Taylor D., Murdoch I.A., Tibby S.M.: The temporal relationship between glucosecorrected serum sodium and neurological status in severe diabetic ketoacidosis. Arch. Dis. Child., 2011; 96: 50–57
203. Bureau M.A., Begin R., Berthiaume Y., Shapcott D., Khoury K., Gagnon N.: Cerebral hypoxia from bicarbonate infusion in diabetic acidosis. J. Pediatr., 1980; 96: 968–973
204. Deeb L.: Development of fatal cerebral edema during outpatient therapy for diabetic ketoacidosis. Pract. Diabetes, 1989; 6: 212–213
205. Glasgow A.M.: Devastating cerebral edema in diabetic ketoacidosis before therapy [letter]. Diabetes Care, 1991; 14: 77–78
206. Couch R.M., Acott P.D., Wong G.W.: Early onset fatal cerebral edema in diabetic keto- acidosis. Diabetes Care, 1991; 14: 78–79
207. Fiordalisi I., Harris G.D., Gilliland M.G.: Prehospital cardiac arrest in diabetic ketoacidemia: why brain swellingmay lead to death before treatment. J. Diabetes Complications, 2002; 16: 214–219
208. Edge J.A.: Cerebral oedema during treatment of diabetic ketoacidosis: are we any nearer finding a cause? Diabetes Metab. Res. Rev., 2000; 16: 316–324
209. Muir A.B., Quisling R.G., Yang M.C., Rosenbloom A.L.: Cerebral edema in childhood diabetic ketoacidosis: natural history, radiographic findings, and early identification. Diabetes Care, 2004; 27: 1541–1546
210. Franklin B., Liu J., Ginsberg-Fellner F.: Cerebral edema and ophthalmoplegia reversed by mannitol in a new case of insulin-dependent diabetes mellitus. Pediatrics, 1982; 69: 87–90
211. Shabbir N., Oberfield S.E., Corrales R., Kairam R., Levine L.S.: Recovery from symptomatic brain swelling in diabetic ketoacidosis. Clin. Pediatr. (Phila), 1992; 31: 570–573
212. Roberts M.D., Slover R.H., Chase H.P.: Diabetic ketoacidosis with intracerebral complications. Pediatr. Diabetes, 2001; 2: 109–114
213. Curtis J.R., Bohn D., Daneman D.: Use of hypertonic saline in the treatment of cerebral edema in diabetic ketoacidosis (DKA). Pediatr. Diabetes, 2001; 2: 191–194
214. Kamat P., Vats A., Gross M., Checchia P.A.: Use of hypertonic saline for the treatment of altered mental status associated with diabetic ketoacidosis. Pediatr. Crit. Care Med., 2003; 4: 239–242
215. Kanter R.K., Oliphant M., Zimmerman J.J., Stuart M.J.: Arterial thrombosis causing cerebral edema in association with diabetic ketoacidosis. Crit. Care Med., 1987; 15: 175–176
216. Roe T.F., Crawford T.O., Huff K.R., Costin G., Kaufman F.R., Nelson M.D. Jr.: Brain infarction in children with diabetic ketoacidosis. J. Diabetes Complications, 1996; 10: 100–108
217. Rosenbloom A.L.: Fatal cerebral infarctions in diabetic ketoacidosis in a child with previously unknown heterozygosity for factor V Leiden deficiency. J. Pediatr., 2004; 145: 561–562
218. Fourtner S.H., Weinzimer S.A., Levitt Katz L.E.: Hyperglycemic hyperosmolar non-ketotic syndrome in children with type 2 diabetes. Pediatr. Diabetes, 2005; 6: 129–135
219. Kronan K., Normal M.E.: Renal and electrolyte emergencies. In: Fleisher G.R., Ludwig S., eds.: Textbook of Emergency Medicine. 4th edn. Philadelphia: Lippincott Williams and Wilkins, 2000
220. Matz R.: Management of the hyperosmolar hyperglycemic syndrome. Am. Fam. Physician, 1999; 60: 1468–1476
221. Delaney M.F., Zisman A., Kettyle W.M.: Diabetic ketoacidosis and hyperglycemic hyperosmolar nonketotic syndrome. Endocrinol. Metab. Clin. North Am., 2000; 29: 683–705, V
222. Mannix R., Tan M.L., Wright R., Baskin M.: Acute pediatric rhabdomyolysis: causes and rates of renal failure. Pediatrics, 2006; 118: 2119–2125
223. Hollander A.S., Olney R.C., Blackett P.R., Marshall B.A.: Fatal malignant hyperthermia-like syndrome with rhabdomyolysis complicating the presentation of diabetes mellitus in adolescent males. Pediatrics, 2003; 111 (6 Pt 1): 1447–1452
224. Carcillo J.A., Tasker R.C.: Fluid resuscitation of hypovolemic shock: acute medicine’s great triumph for children. Intensive Care Med., 2006; 32: 958–961
225. Kilbane B.J., Mehta S., Backeljauw P.F., Shanley T.P., Crimmins N.A.: Approach to management of malignant hyperthermia-like syndrome in pediatric diabetes mellitus. Pediatr. Crit. Care Med., 2006; 7: 169–173
226. Laffel L.M., Wentzell K., Loughlin C., Tovar A., Moltz K., Brink S.: Sick day management using blood 3-hydroxybutyrate (3-OHB) compared with urine ketone monitoring reduces hospital visits in young people with T1DM: a randomized clinical trial. Diabet. Med., 2006; 23: 278–284
227. Laffel L.: Sick-day management in type 1 diabetes. Endocrinol. Metab. Clin. North Am., 2000; 29: 707–723
228. Hoffman W.H., O’Neill P., Khoury C., Bernstein S.S.: Service and education for the insu- lin-dependent child. Diabetes Care, 1978; 1: 285–288
229. Drozda D.J., Dawson V.A., Long D.J., Freson L.S., Sperling M.A.: Assessment of the effect of a comprehensive diabetes management program on hospital admission rates of children with diabetes mellitus. Diabetes Educ., 1990; 16: 389–393
230. Grey M., Boland E.A., Davidson M., Li J., Tamborlane W.V.: Coping skills training for youth with diabetes mellitus has long-lasting effects on metabolic control and quality of life. J. Pediatr., 2000; 137: 107–113
231. Golden M.P., Herrold A.J., Orr D.P.: An approach to prevention of recurrent diabetic ketoacidosis in the pediatric population. J. Pediatr., 1985; 107: 195–200
232. Wolfsdorf J., Glaser N., Sperling M.A.: Diabetic ketoacidosis in infants, children, and adolescents: a consensus statement from the American Diabetes Association. Diabetes Care, 2006; 29: 1150–1159
233. Dunger D., Menon R.K., Sperling M.A.: Ketoacidosis. In: Hochberg Z., ed.: Practical Algorithms in Pediatric Endocrinology. Basel: S. Karger AG, 1999; 104–105














