Wprowadzenie
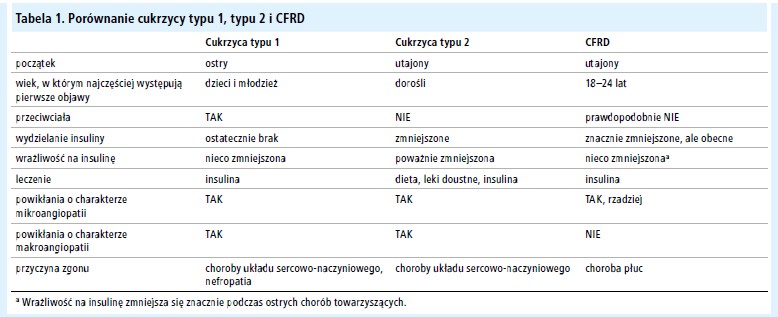
Mukowiscydoza (cystic fibrosis – CF) jest najczęściej występującą u osób rasy kaukaskiej prowadzącą do zgonu chorobą genetyczną, dziedziczoną autosomalnie recesywnie, występującą na całym świecie z częstością 1 na 2500 żywych urodzeń. Cukrzyca stanowi najczęstszą chorobę współistniejącą z mukowiscydozą (cystic fibrosis related diabetes – CFRD).[1,2] Istnieją ważne różnice pomiędzy CFRD a cukrzycą zarówno typu 1, jak i 2, co wymaga indywidualnego podejścia do jej diagnostyki i leczenia (tab. 1).
Definicje
W 1998 roku North American CFRD Consensus Committee wydał zmodyfikowane wytyczne interpretacji OGTT, wskazując na występowanie CFRD z hiperglikemią na czczo oraz bez hiperglikemii na czczo (stężenie glukozy na czczo >=7,0 mmol/l [126 mg/dl] lub <7 mmol/l i stężenie glukozy 2 godziny po obciążeniu >=11,1 mmol/l [200 mg/dl]). Opierano się na przesłankach, że rokowanie w tych dwóch grupach chorych z mukowiscydozą może być różne (E).[2] CFRD stanowi krańcową postać w całym spektrum postępujących zaburzeń tolerancji glukozy u chorych na mukowiscydozę. U nielicznych chorych z mukowiscydozą stężenie glukozy we krwi pozostaje zawsze prawidłowe. Najwcześniejsze zaburzenia mają różnorodną postać, od przemijającej hiperglikemii poposiłkowej, poprzez upośledzoną tolerancję glukozy (IGT), cukrzycę bez hiperglikemii na czczo do cukrzycy z hiperglikemią na czczo. "Prawidłowa" tolerancja glukozy w OGTT nie wyklucza nieprawidłowej glikemii poposiłkowej w warunkach domowych (kiedy spożycie węglowodanów może znacznie przekraczać 75 g) (B).[2]
Przypisanie chorego na mukowiscydozę do konkretnej kategorii diagnostycznej cukrzycy komplikuje fakt, że tolerancja glukozy i insulinooporność są często zmienne u poszczególnych osób. Do typowych dla mukowiscydozy czynników powodujących zmienność metabolizmu glukozy należą: zakażenia i zapalenie układu oddechowego, zwiększony wydatek energetyczny, niedożywienie, niedobór glukagonu oraz zaburzenia ze strony przewodu pokarmowego (upośledzone wchłanianie, upośledzone opróżnianie żołądkowe, zaburzenia motoryki jelit i choroby wątroby).
Częstość występowania
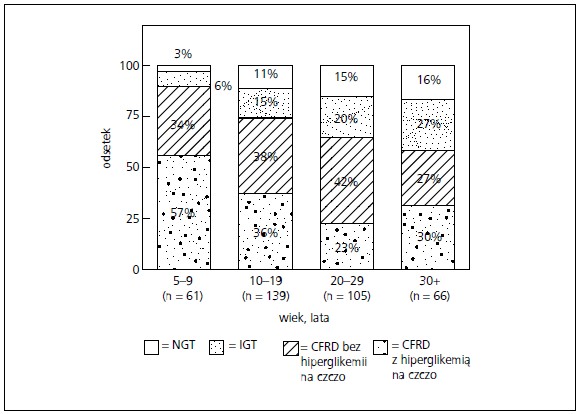
Doniesienia na temat częstości występowania CFRD różnią się w zależności od zastosowanych badań przesiewowych i kryteriów diagnostycznych. W ośrodkach, w których nie prowadzi się powszechnych badań przesiewowych, występowanie może być niedoszacowane. Pomimo iż CFRD może wystąpić w każdym wieku, jej częstość zwiększa się z wiekiem: od 9% w wieku 5–9 lat, do 26% w wieku 10–20 lat (Minnesota)[3], (rys.) oraz do 50% w wieku 30 lat (Dania).[8] Powtarzane OGTT wykazały, że u chorych z mukowiscydozą tolerancja glukozy może zmieniać się z roku na rok (B).[4]
Patofizjologia cukrzycy związanej z mukowiscydozą
Genetyka CFRD i jej związek z rodzajem mutacji powodującej mukowiscydozę
CFRD występuje głównie u osób z mutacjami powodującymi mukowiscydozę o najcięższym przebiegu, u których występuje też zewnątrzwydzielniczna niewydolność trzustki (C).[5-7] Nie wykazano związku ze znanymi genami podatności na cukrzycę typu 1, takimi jak HLA klasy II8 (B) czy gen insuliny VNTR[5] (C), ale opisano możliwy związek pomiędzy CFRD a genami podatności na cukrzycę typu 1 związanymi z czynnikami zapalnymi, takimi jak czynnik martwicy nowotworów (tumor necrosis factor – TNF)[8] (B), białko szoku termicznego (heat shock protein – HSP)[8,9] (B, C) oraz genami podatności na cukrzycę typu 2, takimi jak gen kalpainy 10[10] (C).
Zaburzenia czynności trzustki
Nieprawidłowe działanie kanałów chlorkowych w mukowiscydozie jest przyczyną produkcji lepkiej wydzieliny powodującej zatykanie przewodów, prowadzące do upośledzenia zewnątrzwydzielniczej części trzustki, postępującego włóknienia i stłuszczenia. Skutkuje to zakłóceniem i zniszczeniem architektury wysp, prowadzącym do utraty wydzielniczej funkcji komórek β, α oraz komórek produkujących polipeptydy trzustkowe (B).[11-13] Dysfunkcja komórek β w mukowiscydozie nie jest związana z autoimmunizacją, poza kilkoma pojedynczymi opisami przypadków osób, u których stwierdzono obecność autoprzeciwciał w przebiegu CFRD.[14]
Znaczenie niedoboru insuliny
Pierwotnym zaburzeniem w CFRD jest ciężki, ale nie całkowity niedobór insuliny. Praktycznie wszyscy chorzy z niewydolnością zewnątrzwydzielniczą trzustki w przebiegu mukowiscydozy, niezależnie od tego, czy chorują na cukrzycę czy nie, wykazują objawy niewydolności komórek β (A).[4,15-17] Stężenia insuliny i peptydu C na czczo są prawidłowe, ale występuje opóźnienie i zmniejszenie szczytu wydzielania insuliny w trakcie standardowego OGTT (B).[18] Ten efekt jest silniej wyrażony przy pogarszaniu się kontroli glikemii (B, C).[18-21] Opóźnione wydzielanie insuliny w OGTT jest związane z utratą pierwszej fazy wydzielania insuliny, która występuje nawet u tych chorych z mukowiscydozą, u których tolerancja glukozy jest prawidłowa (B).[22] W mukowiscydozie upośledzone jest także wydzielanie innych hormonów trzustkowych, szczególnie następuje zanik reaktywnego wydzielania glukagonu (B).[18,22]
Znaczenie insulinooporności
U chorych z mukowiscydozą bez cukrzycy wrażliwość na insulinę jest zmienna[21,23-26] (B)[17,27] (B). Podczas gdy u większości tych chorych w stabilnym stanie zdrowia wrażliwość na insulinę jest zachowana, zakażenie i zapalenie nasilają insulinooporność (B).[28] U chorych z mukowiscydozą i cukrzycą występuje insulinooporność spowodowana zarówno zmniejszonym wychwytem glukozy przez tkanki obwodowe, jak i słabym hamowaniem przez insulinę wątrobowej produkcji glukozy (B).[26,27] Insulinooporność może się gwałtownie nasilać podczas zaostrzeń infekcji (E).
Objawy kliniczne CFRD
CFRD rozwija się skrycie, a chorzy przez wiele lat mogą nie wykazywać objawów. Objawy CFRD wymieniono w tabeli 2. Cukrzycowa kwasica ketonowa występuje rzadko, najprawdopodobniej z powodu zachowania wydzielania endogennej insuliny lub równoczesnego upośledzenia wydzielania glukagonu (B).[22,29,30] Pierwsze objawy CFRD występują zwykle w sytuacjach nasilających insulinooporność, takich jak: ostre zapalenie płuc, przewlekła ciężka choroba płuc, leczenie glikokortykosteroidami, przyjmowanie dużych ilości węglowodanów (podaż doustna, dożylna, przez sondę żołądkową lub gastrostomię przezskórną) oraz w związku z leczeniem immunosupresyjnym po przeszczepieniu narządów. CFRD występuje częściej u osób z chorobą wątroby w przebiegu mukowiscydozy (C).[31] Hipoglikemia u osób z chorobą wątroby występuje częściej i wiąże się z większym zagrożeniem. Jeśli nie stwierdza się choroby wątroby, hipoglikemia występuje wyłącznie u osób niedożywionych i w bardzo młodym wieku. Hipoglikemia reaktywna może występować u chorych na mukowiscydozę z upośledzoną tolerancją glukozy i można ją złagodzić, równomiernie rozkładając podaż węglowodanów w czasie (E).

Przeżywalność i rokowanie
Zwiększona śmiertelność u chorych z CFRD
Wystąpienie CFRD jest związane z gorszą funkcją płuc, gorszym stanem odżywienia oraz skróceniem przeżycia w porównaniu z chorymi na mukowiscydozę bez cukrzycy (A–C).[31-38] W jednym badaniu retrospektywnym, które objęło 448 chorych z mukowiscydozą obserwowanych przez 10 lat, wykazano, że 30. roku życia dożyło 25% chorych z CFRD w porównaniu z 60% chorych bez CFRD (C).[32] Ostatnio udokumentowano znaczne różnice przeżywalności chorych z CFRD związane z płcią, mediana przeżycia wyniosła 47–49 lat u mężczyzn i tylko 31 lat u kobiet (B).[39] Takich związanych z płcią różnic nie stwierdzono z kolei w przeprowadzonym niedawno we Francji prospektywnym badaniu kohortowym obejmującym 237 dzieci chorych na mukowiscydozę (B).[29]
Zwiększona chorobowość w stanie przedcukrzycowym
Utajone pogarszanie się stanu klinicznego może mieć miejsce w okresie 2–6 lat przed rozpoznaniem CFRD (B).[32,34-37] Pogorszenie funkcji płuc koreluje ze stopniem niedoboru podstawowego wydzielania insuliny.[41] Znany jest związek pomiędzy katabolizmem białek, niedożywieniem a zgonem chorych na mukowiscydozę. Brak silnego anabolicznego działania insuliny (wpływ niedoboru insuliny na stan odżywienia i metabolizm) może mieć w mukowiscydozie większe znaczenie niż metaboliczny wpływ hiperglikemii (E).
Powikłania o charakterze mikroangiopatii
U chorych na CFRD opisywano powikłania o charakterze mikroangiopatii, które niekiedy powodują znaczną chorobowość (C).[40-42] W badaniu przeprowadzonym w Danii opisano występowanie retinopatii u 36% chorych z CFRD trwającą ponad 10 lat (B).[43] W innym dużym badaniu obserwacyjnym u chorych z CFRD z hiperglikemią na czczo po 10 latach trwania choroby wykazano obecność mikroalbuminurii u 14%, retinopatii u 16%, neuropatii u 55% a gastropatii u 50% chorych. Powikłania o charakterze mikroangiopatii rzadko występowały przed upływem 10 lat trwania CFRD z hiperglikemią na czczo. Nie wykazano zaburzeń o charakterze mikroangiopatii u chorych z CFRD bez hiperglikemii na czczo przed upływem 14 lat trwania choroby.[44] Dotychczas nie opisano występowania powikłań o charakterze makroangiopatii u chorych z CFRD.
Badania przesiewowe w kierunku CFRD
Ważne znaczenie ma wykrywanie CFRD u chorych przed wystąpieniem objawów, ponieważ jej początek często przebiega w sposób skryty. Stosuje się następujące metody badań w kierunku CFRD: oznaczenie hemoglobiny A1c (HbA1c), OGTT, oznaczenie glikemii na czczo lub przygodnej oraz ciągłe monitorowanie glikemii (continuous glucose monitoring – CGM).
HbA1c jako narzędzie diagnostyczne
Wykazano brak wiarygodności oznaczenia HbA1c w diagnostyce CFRD (B).[4,19,32,45] Odsetek HbA1c często jest prawidłowy, niezależnie od nasilenia hiperglikemii, a tylko u 16% chorych stwierdza się zwiększone stężenie HbA1c w chwili postawienia rozpoznania cukrzycy (B).[4]
OGTT
OGTT jest w wielu ośrodkach standardowym badaniem przesiewowym w kierunku CFRD4 (A)46 (E). Cukrzycę bez hiperglikemii na czczo można zdiagnozować wyłącznie za pomocą OGTT. Pomiar stężenia insuliny co pół godziny w trakcie testu może być użyteczny klinicznie do określenia stopnia niedoboru insuliny (E).
Glikemia przygodna i glikemia na czczo w diagnostyce CFRD
Chociaż hiperglikemia jest kryterium diagnostycznym cukrzycy, prawidłowe stężenie glukozy na czczo nie wyklucza rozpoznania cukrzycy w mukowiscydozie (A).
Ciągłe monitorowanie glikemii (CGM)
W badaniach naukowych CGM może wykryć upośledzoną tolerancję glukozy wcześniej niż OGTT, ale znaczenie kliniczne testu ciągle pozostaje przedmiotem badań. Potwierdzono wartość CGM u dzieci i młodzieży z mukowiscydozą.[47-51] CGM może pomóc w postawieniu rozpoznania CFRD, jeśli analizuje się je wspólnie z wynikiem OGTT oraz przebiegiem klinicznym choroby (C).[16,48,49]
Podejrzenie CFRD na podstawie objawów klinicznych
W przypadku gdy wynik OGTT jest prawidłowy lub graniczny, a na podstawie objawów klinicznych podejrzewa się cukrzycę, dodatkowych użytecznych informacji mogą dostarczyć okresowe pomiary glikemii w domu (przed posiłkiem i 2 godziny po posiłku oraz w połowie nocnego karmienia sondą żołądkową) lub CGM (E).
Leczenie CFRD
Leczenie żywieniowe
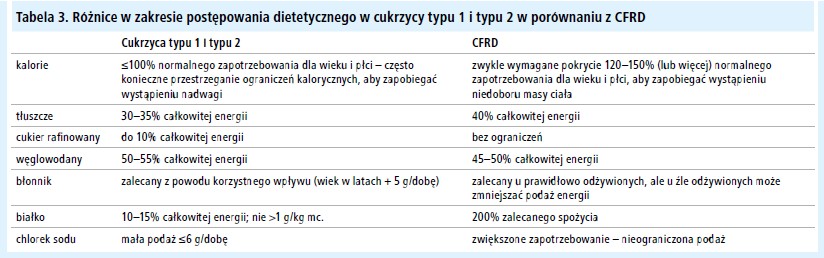
Ważne znaczenie w mukowisydozie ma dieta wysokokaloryczna, bogata w tłuszcze (A).[52] Z tego powodu ograniczenie podaży kalorii jest przeciwwskazane. W tabeli 3 przedstawiono porównanie diety w CFRD i typowej diety cukrzycowej.
Leczenie insuliną
Insulina jest jedynym lekiem zalecanym do stosowania w leczeniu CFRD (E).[2] Insulinoterapia może pomóc w stabilizacji funkcji płuc i poprawić stan odżywienia chorych z CFRD (C).[34,36,37,53] Dotychczas nie ma ostatecznych danych na temat korzyści leczenia insuliną u dzieci i młodzieży z mukowiscydozą i łagodniejszymi postaciami upośledzonej tolerancji glukozy, chociaż badania przeprowadzone na małych grupach wykazały podobną korzyść (C).[16,54]
Wybór schematu dawkowania zależy od indywidualnych potrzeb i charakterystyki chorego. Standardowa insulinoterapia według schematu wielokrotnych wstrzyknięć zapewnia podstawowe stężenie insuliny i jej ciągłe działanie anaboliczne. Insulina krótko działająca kontroluje epizody hiperglikemii poposiłkowej i zapewnia elastyczność w przypadku zmiennych godzin spożywania posiłków (B).[55] Intensywną insulinoterapię można też skutecznie prowadzić z użyciem osobistej pompy insulinowej (E).[56,57]
Insulina izofanowa podawana 2 razy na dobę z insuliną krótko działającą lub bez niej może być stosowana, ale może nie być optymalnym sposobem leczenia, ponieważ nie pozwala na elastyczne spożywanie posiłków w związku z występowaniem nudności lub jadłowstrętu albo koniecznością stosowania innych leków, zwłaszcza w godzinach porannych.
W czasie podejmowania decyzji o schemacie leczenia insuliną u danego pacjenta należy przeanalizować dotychczasowe leczenie i stosowanie się do zaleceń.
U chorych, u których stosowane jest żywienie dojelitowe w godzinach nocnych, zapotrzebowanie na insulinę w nocy jest większe. Zaleca się monitorowanie glikemii w trakcie karmienia, aby umożliwić odpowiednie dostosowanie dawki insuliny (tab. 4).

Doustne leki przeciwcukrzycowe
Doustne leki przeciwcukrzycowe obecnie nie są zalecane w CFRD (E).[2] W niedawnym przeglądzie Cochrane podkreślono, że nie ma badań z randomizacją dotyczących tego zagadnienia (A).[58] W badaniu eksperymentalnym wykazano, że pobudzający wydzielanie insuliny repaglinid zwiększał stężenie endogennej insuliny, ale był mniej skuteczny od szybko działającego analogu insuliny w regulacji poposiłkowej hiperglikemii (B).[59] Istnieją obawy o występowanie hipoglikemii podczas stosowania pochodnych sulfonylomocznika u chorych z mukowiscydozą[60] (B), (E).
Leki zmniejszające insulinooporność stosowane w monoterapii raczej nie są skuteczne w CFRD, ponieważ insulinooporność nie jest głównym czynnikiem etiologicznym. Niepożądane działania metforminy na przewód pokarmowy, powodujące nudności, biegunkę i dyskomfort w jamie brzusznej, nie są akceptowane przez większość chorych z mukowiscydozą (C).[61] Ostatnio wykazano, że stosowanie pochodnych tiazolidynodionu jest związane z osteoporozą. Spowolnienie opróżniania żołądkowego wywoływane przez leki działające na układ inkretynowy ogranicza ich przydatność w mukowiscydozie.
CFRD bez hiperglikemii na czczo i mukowiscydoza z upośledzoną tolerancją glukozy
Podczas gdy w CFRD z hiperglikemią na czczo leczenie insuliną zostało kilka lat temu zaakceptowane jako standard, leczenie lżejszych zaburzeń tolerancji glukozy jest bardziej kontrowersyjne. Wyniki niedawno przeprowadzonego wieloośrodkowego badania z randomizacją i z grupą kontrolną otrzymującą placebo wykazały, że podawanie insuliny przed posiłkami powodowało odwrócenie trendu przewlekłego zmniejszania się masy ciała u dorosłych z CFRD bez hiperglikemii na czczo, a działanie to utrzymało się w ciągu rocznego leczenia.[62] Z tego powodu obecnie zaleca się leczenie insuliną wszystkich chorych z CFRD z hiperglikemią na czczo lub bez hiperglikemii na czczo (B). Obecnie nie ma wystarczających danych, aby opracować zalecenia dla chorych z IGT lub chorych, u których tolerancja glukozy w OGTT jest prawidłowa, a okresowo występują bezobjawowe epizody hiperglikemii stwierdzane w pomiarach wykonywanych w domu.
Postępowanie z chorym na CFRD w szpitalu
Podczas ostrej choroby towarzyszącej u dzieci i młodzieży z mukowiscydozą występuje zwiększone ryzyko rozwoju hiperglikemii (E).[2] Podczas gdy dane z innych populacji sugerują, że intensywna insulinoterapia może być korzystna w takich okolicznościach, w żadnym badaniu nie oceniano korzyści wynikających z utrzymania normoglikemii u hospitalizowanych chorych z mukowiscydozą. Zapotrzebowanie na insulinę podczas ostrej choroby towarzyszącej może być duże: niektórzy chorzy z CFRD wymagają nawet czterokrotnego zwiększenia dawki podstawowej (E). Dawka insuliny musi być szybko zmniejszana w okresie zdrowienia, aby uniknąć hipoglikemii, a u wielu chorych z mukowiscydozą stężenie glukozy we krwi powraca do normy po ustąpieniu choroby (E).[2]
Zalecenia
Kryteria diagnostyczne CFRD
Diagnostyka CFRD
– wystąpienie objawów cukrzycy, takich jak wymienione w tabeli 2
– zaostrzenia związane z zakażeniem
– leczenie glikokortykosteroidami stosowanymi ogólnie
– rozpoczęcie uzupełniającego żywienia przez sondę jelitową
– stan przed zabiegami i po dużych zabiegach chirurgicznych
– objawy hipoglikemii
– ciąża – wymaga szczególnej uwagi (C).[63]
Leczenie CFRD
Decyzję o leczeniu należy podjąć na podstawie analizy stężeń glukozy we krwi oraz uwzględnić wpływ leczenia na stan ogólny chorego.
CFRD z hiperglikemią na czczo
CFRD bez hiperglikemii na czczo
Obecnie zalecanym lekiem jest insulina (B).
Mukowiscydoza z IGT
Piśmiennictwo
1. CFF 2001 Cystic Fibrosis Foundation Patient Registry Annual Data Report In: CF Foundation Bethesda, Maryland
2. Moran A., Hardin D., Rodman D., Allen H.F., Beall R.J., Borowitz D., Brunzell C., Campbell P.W. 3rd, Chesrown S.E., Duchow C., Fink R.J., Fitzsimmons S.C., Hamilton N., Hirsch I., Howenstine M.S., Klein D.J., Madhun Z, Pencharz Pb, Quittner A.L., Robbins M.K., Schindler T., Schissel K., Schwarzenberg S.J., Stallings V.A., Zipf W.B., et al.: Diagnosis, screening and management of cystic fibrosis related diabetes mellitus: a consensus conference report. Diabetes Res. Clin. Pract., 1999; 45: 61–73
3. Moran A., Doherty L., Wang X., Thomas W.: Abnormal glucose metabolism in cystic fibrosis. J. Pediatr., 1998; 133: 10–17
4. Lanng S., Hansen A., Thorsteinsson B., Nerup J., Koch C.: Glucose tolerance in patients with cystic fibrosis: five year prospective study. BMJ, 1995; 311: 655–659
5. O'riordan S.G.A., Ennis S., George S., Hand E., Costigan C., Murphy N., Roche E., Hoey H.: Genetics of cystic fibrosis related diabetes and non diabetes. Horm. Res., 2007; 68 (Suppl.1): 44–69, PO2-345 Proceedings from The European Society for Paediatric Endocrinology (ESPE), 2007 Helsinki 978-3-8055-8327-5: 1–283
6. Rosenecker J., Eichler I., Kuhn L., Harms H.K., Von Der Hardt H.: Genetic determination of diabetes mellitus in patients with cystic fibrosis. J. Pediatr., 1995; 127: 441–443
7. Koch C., Cuppens H., Rainisio M., Madessani U., Harms H., Hodson M., Mastella G., Navarro J., Strandvik B., Mckenzie S.: European Epidemiologic Registry of Cystic Fibrosis (ERCF): comparison of major disease manifestations between patients with different classes of mutations. Pediatr. Pulmonol., 2001; 31: 1–12
8. Lanng S., Thorsteinsson B., Pociot F., et al.: Diabetes Mellitus in cystic fibrosis: genetic and immunological markers. Acta Paediatr. Scand., 1993; 82
9. Jensen P., Johansen H.K., Carmi P., Hoiby N., Cohen I.R.: Autoantibodies to pancreatic hsp60 precede the development of glucose intolerance in patients with cystic fibrosis. J. Autoimmun., 2001; 17: 165–172
10. Derbel S., Doumaguet C., Hubert D., Mosnierpudar H., Grabar S., Chelly J., Bienvenu T.: Calpain 10 and development of diabetes mellitus in cystic fibrosis. J. Cyst. Fibros., 2006; 5: 47–51
11. Couce M., O'brien T.D., Moran A., Roche P.C., Butler P.C.: Diabetes mellitus in cystic fibrosis is characterized by islet amyloidosis. J. Clin. Endocrinol. Metab., 1996; 81: 1267–1272
12. Lohr M., Goertchen P., Nizze H., Gould N.S., Gould V.E., Oberholzer M., Heitz P.U., Kloppel G.: Cystic fibrosis associated islet changes may provide a basis for diabetes. An immunocytochemical and morphometrical study. Virchows Arch. A Pathol. Anat. Histopathol., 1989; 414: 179–185
13. Iannucci A., Mukai K., Johnson D., Burke B.: Endocrine pancreas in cystic fibrosis: an immunohistochemical study. Hum. Pathol., 1984; 15: 278–284
14. Lanng S., Thorsteinsson B., Pociot F., Marshall M.O., Madsen H.O., Schwartz M., Nerup J., Koch C.: Diabetes mellitus in cystic fibrosis: genetic and immunological markers. Acta Paediatr., 1993; 82; 150–154
15. Dobson L., Sheldon C., Hattersley A.T.: Understanding cystic-fibrosis-related diabetes: best thought of as insulin deficiency? J. R. Soc. Med., 2004; 97 (suppl. 44): 26–35
16. Dobson L., Sheldon C.D., Hattersley A.T.: Conventional measures underestimate glycaemia in cystic fibrosis patients. Diabet. Med., 2004; 21: 691–696
17. Holl R.W., Heinze E., Wolf A., Rank M., Teller W.M.: Reduced pancreatic insulin release and reduced peripheral insulin sensitivity contribute to hyperglycaemia in cystic fibrosis. Eur. J. Pediatr., 1995; 154: 356–361
18. Lanng S., Thorsteinsson B., Roder M.E., Orskov C., Holst J.J., Nerup J., Koch C.: Pancreas and gut hormone responses to oral glucose and intravenous glucagon in cystic fibrosis patients with normal, impaired, and diabetic glucose tolerance. Acta Endocrinol. (Copenh), 1993; 128: 207–214
19. De Schepper J., Dab I., Derde M.P., Loeb H.: Oral glucose tolerance testing in cystic fibrosis: correlations with clinical parameters and glycosylated haemoglobin determinations. Eur. J. Pediatr., 1991; 150: 403–406
20. Hamdi I., Green M., Shneerson J.M., Palmer C.R., Hales C.N.: Proinsulin, proinsulin intermediate and insulin in cystic fibrosis. Clin. Endocrinol. (Oxf.), 1993; 39: 21–26
21. Yung B., Noormohamed F.H., Kemp M., Hooper J., Lant A.F., Hodson M.E.: Cystic fibrosisrelated diabetes: the role of peripheral insulin resistance and beta-cell dysfunction. Diabet. Med., 2002; 19: 221–226
22. Moran A., Diem P., Klein D.J., Levitt M.D., Robertson R.P.: Pancreatic endocrine function in cystic fibrosis. J. Pediatr., 1991; 118: 715–723
23. Ahmad T., Nelson R., Taylor R.: Insulin sensitivity and metabolic clearance rate of insulin in cystic fibrosis. Metabolism, 1994; 43: 163–167
24. Cucinotta D., De Luca F., Gigante A., Arrigo T., Di Benedetto A., Tedeschi A., Lombardo F., Romano G., Sferlazzas C.: No changes of insulin sensitivity in cystic fibrosis patients with different degrees of glucose tolerance: an epidemiological and longitudinal study. Eur. J. Endocrinol., 1994; 130: 253–258
25. Lombardo F., De Luca F., Rosano M., Sferlazzas C., Lucanto C., Arrigo T., Messina M.F., Crisafulli G., Wasniewska M., Valenzise M., Cucinotta D.: Natural history of glucose tolerance, beta-cell function and peripheral insulin sensitivity in cystic fibrosis patients with fasting euglycemia. Eur. J. Endocrinol., 2003; 149: 53–59
26. Moran A., Pyzdrowski K.L., Weinreb J., Kahn B.B., Smith S.A., Adams K.S., Seaquist E.R.: Insulin sensitivity in cystic fibrosis. Diabetes, 1994; 43: 1020–1026
27. Hardin D.S., Leblanc A., Lukenbough S., Seilheimer D.K.: Insulin resistance is associated with decreased clinical status in cystic fibrosis. J. Pediatr., 1997; 130: 948–956
28. Hardin D.S., Leblanc A., Marshall G., Seilheimerd K.: Mechanisms of insulin resistance in cystic fibrosis. Am. J. Physiol. Endocrinol. Metab., 2001; 281: E1022–E1028
29. Bismuth E., Laborde K., Taupin P., Velho G., Ribault V., Jennane F., Grasset E., Sermet I., De Blic J., Lenoir G., Robert J.J.: Glucose tolerance and insulin secretion, morbidity, and death in patients with cystic fibrosis. J. Pediatr., 2008; 152: 540–545, 545.e1
30. Lanng S., Thorsteinsson B., Erichsen G., Nerup J., Koch C.: Glucose tolerance in cystic fibrosis. Arch. Dis. Child., 1991; 66: 612–616
31. Marshall B.C., Butler S.M., Stoddard M., Moran A.M., Liou T.G., Morgan W.J.: Epidemiology of cystic fibrosis-related diabetes. J. Pediatr. 2005; 146: 681–687
32. Finkelstein S.M., Wielinski C.L., Elliott G.R., Warwick W.J., Barbosa J., Wu S.C., Klein D.J.: Diabetes mellitus associated with cystic fibrosis. J. Pediatr., 1988; 112: 373–377
33. Koch C., Rainisio M., Madessani U., Harms H.K., Hodson M.E., Mastella G., Mckenzie S.G., Navarro J., Strandvik B.: Presence of cystic fibrosis-related diabetes mellitus is tightly linked to poor lung function in patients with cystic fibrosis: data from the European Epidemiologic Registry of Cystic Fibrosis. Pediatr. Pulmonol., 2001; 32: 343–350
34. Lanng S., Thorsteinsson B., Nerup J., Koch C.: Influence of the development of diabetes mellitus on clinical status in patients with cystic fibrosis. Eur. J. Pediatr., 1992; 151: 684–687
35. Milla C.E., Warwick W.J., Moran A.: Trends in pulmonary function in patients with cystic fibrosis correlate with the degree of glucose intolerance at baseline. Am. J. Respir. Crit. Care Med., 2000; 162: 891–895
36. Nousia-Arvanitakis S., Galli-Tsinopoulou A., Karamouzis M.: Insulin improves clinical status of patients with cystic-fibrosis-related diabetes mellitus. Acta Paediatr., 2001; 90: 515–519
37. Rolon M.A., Benali K., Munck A., Navarro J., Clement A., Tubiana-Rufi N., Czernichow P., Polak M.: Cystic fibrosis-related diabetes mellitus: clinical impact of prediabetes and effects of insulin therapy. Acta Paediatr., 2001; 90: 860–867
38. Rosenecker J., Hofler R., Steinkamp G., Eichler I., Smaczny C., Ballmann M., Posselt H.G., Bargon J., Von Der Hardt H.: Diabetes mellitus in patients with cystic fibrosis: the impact of diabetes mellitus on pulmonary function and clinical outcome. Eur. J. Med. Res., 2001; 6: 345–350
39. Milla C. E., Billings J., Moran A.: Diabetes is associated with dramatically decreased survival in female but not male subjects with cystic fibrosis. Diabetes Care, 2005; 28: 2141–2144
40. Sullivan M.M., Denning C.R.: Diabetic microangiopathy in patients with cystic fibrosis. Pediatrics, 1989; 84: 642–647
41. Lanng S., Thorsteinsson B., Lund-Andersen C., Nerup J., Schiotz P.O., Koch C.: Diabetes mellitus in Danish cystic fibrosis patients: prevalence and late diabetic complications. Acta Paediatr., 1994; 83: 72–77
42. Yung B., Landers A., Mathalone B., Gyi Km, Hodson M.E.: Diabetic retinopathy in adult patients with cystic fibrosis-related diabetes. Respir. Med., 1998; 92: 871–872
43. Andersen H.U., Lanng S., Pressler T., Laugesen C.S., Mathiesen E.R.: Cystic fibrosisrelated diabetes – The presence of microvascular diabetes complications. Diabetes Care, 2006; 29: 2660–2663
44. Schwarzenberg S.J., Thomas W., Olsen T.W., Grover T., Walk D., Milla C., Moran A.: Microvascular Complications in Cystic Fibrosis Related Diabetes. Diabetes Care, 2007; 30 (5): 1056–1061
45. De Luca F., Arrigo T., Conti Nibali S., Sferlazzas C., Gigante A., Di Cesare E., Cucinotta D.: Insulin secretion, glycosylated haemoglobin and islet cell antibodies in cystic fibrosis children and adolescents with different degrees of glucose tolerance. Horm. Metab. Res., 1991; 23: 495–498
46. Littlewood J., Hodson M., et al.: Management of cystic fibrosis related diabetes mellitus – Report of the UK Cystic Fibrosis Trust Diabetes Working Group. Bromley Kent, UK: Cystic Fibrosis Trust, 2004
47. Jefferies C., Solomon M., Perlman K., Sweezey N., Daneman D.: Continuous glucose monitoring in adolescents with cystic fibrosis. J. Pediatr., 2005; 147: 396–398
48. O'Riordan S., Hoey H., George S., Costigan C.: Can continuous glucose monitoring (CGMS) enhance the detection of CFRD in 167 cystic fibrosis children. Diabetes Care, 2006; 72: A17
49. O'Riordan S., Roche E., George S., Hoey H., Costigan C.: Continuous glucose monitoring enhances the detection of cystic fibrosis related diabetes in children with cystic fibrosis. Diabetologia, 2007; 50 (suppl 1. OP 32, 0190): S1–S538
50. Dobson L., Sheldon C.D., Hattersley A.T.: Validation of interstitial fluid continuous glucose monitoring in cystic fibrosis. Diabetes Care, 2003; 26: 1940–1941
51. O'Riordan S.M., Hindmarsh P., Hill N.R, Matthews D.R., George S., Greally P., Canny G., Slattery D., Murphy N., Roche E., Costigan C., Hoey H.: Validation of continuous glucose monitoring in children and adolescents with cystic fibrosis A prospective cohort study. Diabetes Care, 2009; 32: 1020–1022
52. O'Riordan S.M., Hindmarsh P., Hill N.R., Matthews D.R., George S., Greally P., Canny G., Slattery D., Murphy N., Roche E., Costigan C., Hoey H.: Validation of continuous glucose monitoring in children and adolescents with cystic fibrosis: A prospective cohort study. Diabetes Care, 2009; 32: 1020–1022
53. Borowitz D., Baker R.D., Stallings V.: Consensus report on nutrition for pediatric patients with cystic fibrosis. J. Pediatr. Gastroenterol. Nutr., 2002; 35: 246–259
54. Reisman J., Corey M., Canny G., Levison H.: Diabetes mellitus in patients with cystic fibrosis: effect on survival. Pediatrics, 1990; 86: 374–377
55. Dobson L., Hattersley A.T., Tiley S., Elworthy S., Oades P.J., Sheldon C.D.: Clinical improvement in cystic fibrosis with early insulin treatment. Arch. Dis. Child., 2002; 87: 430–431
56. Grover P., Thomas W., Moran A.: Glargine versus NPH insulin in cystic fibrosis related diabetes. J. Cyst. Fibros., 2008; 7: 134–136
57. Sulli N., Bertasi S., Zullo S., Shashaj B.: Use of continuous subcutaneous insulin infusion in patients with cystic fibrosis related diabetes: Three case reports. J. Cyst. Fibros., 2007; 6 (3): 237–240
58. Sulli N., Shashaj B.: Continuous subcutaneous insulin infusion in children and adolescents with diabetes mellitus: Decreased HbA1c with low risk of hypoglycemia. J. Pediatr. Endocrinol. Metab., 2003; 16: 393–399
59. Onady G.M., Stolfi A.: Insulin and oral agents for managing cystic fibrosis-related diabetes. Cochrane Database Syst. Rev., 2005; 20 (3): CD004730
60. Moran A., Phillips J., Milla C.: Insulin and glucose excursion following premeal insulin lispro or repaglinide in cystic fibrosis-related diabetes. Diabetes Care, 2001; 24: 1706–1710
61. Sheppard D.N., Welsh M.J.: Effect of ATP-Sensitive K+ Channel Regulators on Cystic- Fibrosis Transmembrane Conductance Regulator Chloride Currents. J. Gen. Physiol., 1992; 100: 573–591
62. Moran A., Pekow P., Grover P., Zorn M., Slovis B., Pilewski J., Tullis E., Liou T.G., Allen H.: Insulin therapy to improve BMI in cystic fibrosis-related diabetes without fasting hyperglycemia: results of the CFRDT trial. Diabetes Care, 2009; 32 (10): 1783–1788
63. Rosenecker J., Eichler I., Barmeier H., Von Der Hardt H.: Diabetes mellitus and cystic fibrosis: comparison of clinical parameters in patients treated with insulin versus oral glucose-lowering agents. Pediatr. Pulmonol., 2001; 32: 351–355
64. Moran A., Pekow P., Grover P., Zorn M., Slovis B., Pilewski J., Tullis E., Liou T.G., Allen H.: Insulin therapy to improve BMI in cystic fibrosis related diabetes without fasting hyperglycemia: results of the CFRDT trial. Diabetes Care, 2009; 32 (10): 1783–1788
65. Hardin D.S, Rice J., Cohen R.C., Ellis K.J., Nick J.A.: The metabolic effects of pregnancy in cystic fibrosis. Obstet. Gynecol., 2005; 106: 367–375
Konflikt interesów
Autorzy nie zgłosili konfliktu interesów.