Definicja
Mianem cukrzycy określa się grupę chorób metabolicznych charakteryzujących się przewlekłą hiperglikemią wynikającą z zaburzenia wydzielania i/lub działania insuliny. Zaburzenia metabolizmu węglowodanów, lipidów i białek stwierdzane w cukrzycy są konsekwencją niedostatecznego działania insuliny w tkankach docelowych. Po stwierdzeniu ciał ketonowych we krwi lub moczu należy natychmiast rozpocząć leczenie z uwagi na możliwość szybkiego rozwoju cukrzycowej kwasicy ketonowej.
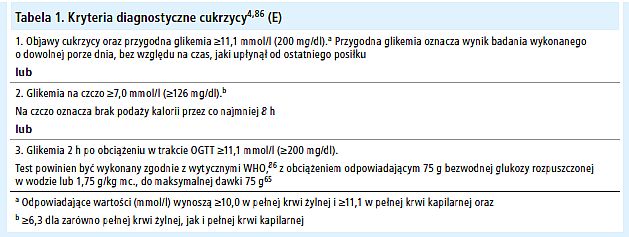
Kryteria diagnostyczne cukrzycy u dzieci i młodzieży
Kryteria diagnostyczne cukrzycy opierają się na pomiarach stężenia glukozy we krwi oraz obecności lub braku objawów (E).[4,86] Istnieją trzy sposoby rozpoznania cukrzycy. W każdym przypadku, w którym występuje wątpliwa hiperglikemia, konieczne jest potwierdzenie rozpoznania w kolejnym dniu, za pomocą jednej z trzech metod wymienionych w tabeli 1.
Upośledzona tolerancja glukozy (impaired glucose tolerance – IGT) oraz nieprawidłowa glikemia na czczo (impaired fasting glycemia – IFG)

Kategorie stężenia glukozy w osoczu na czczo (fasting plasma glucose – FPG) definiowane są w następujący sposób:
Odpowiednie kategorie w przypadku wykonania OGTT są następujące:
Patogeneza cukrzycy typu 1
W niektórych populacjach pojawienie się autoprzeciwciał typowych dla cukrzycy wiązano z zakażeniem enterowirusami,[48,69] a enterowirusy wykryto w wyspach trzustkowych osób chorych na cukrzycę.[16,67,88]
Epidemiologia cukrzycy typu 1
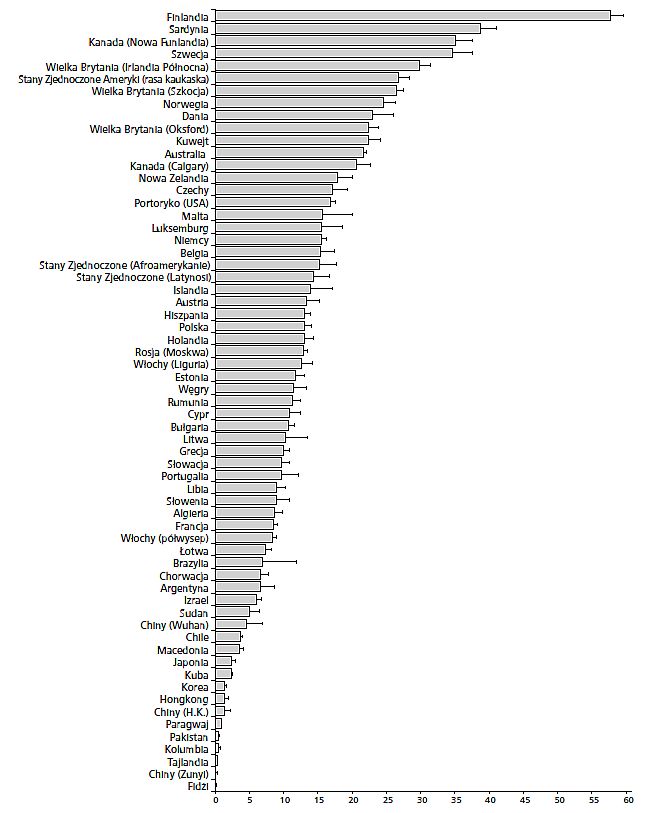
Rys. Porównanie średniej rocznej zapadalności na cukrzycę typu 1 (w grupie wiekowej 0–14 lat) w różnych krajach świata
Klasyfikacja
Klasyfikacja etiologiczna rekomendowana przez ADA (E)4 oraz komitet ekspertów WHO do spraw klasyfikacji i diagnostyki cukrzycy (E)[86] została z niewielkimi zmianami przedstawiona w tabeli 2.
Klasyfikacja typów cukrzycy
Rozróżnienie pomiędzy cukrzycą typu 1, typu 2 i cukrzycą monogenową ma istotne konsekwencje zarówno dla decyzji terapeutycznych, jak i edukacji chorych. Jednakże bez względu na typ cukrzycy, dziecko z objawami ciężkiej hiperglikemii na czczo, znacznymi zaburzeniami metabolicznymi oraz ketonemią wymaga początkowo insulinoterapii w celu odwrócenia nieprawidłowości metabolicznych.[72]
Możliwość występowania innego typu cukrzycy u dziecka należy wziąć pod uwagę w następujących sytuacjach:
– autosomalny dominujący typ dziedziczenia cukrzycy w rodzinie
– choroby współistniejące, takie jak głuchota, zanik nerwu wzrokowego lub cechy znanych zespołów chorobowych
– znacznego stopnia insulinooporność albo niewielkie lub zerowe zapotrzebowanie na insulinę poza okresem częściowej remisji
– wywiad potwierdzający narażenie na leki toksyczne dla komórek β lub powodujące insulinooporność.
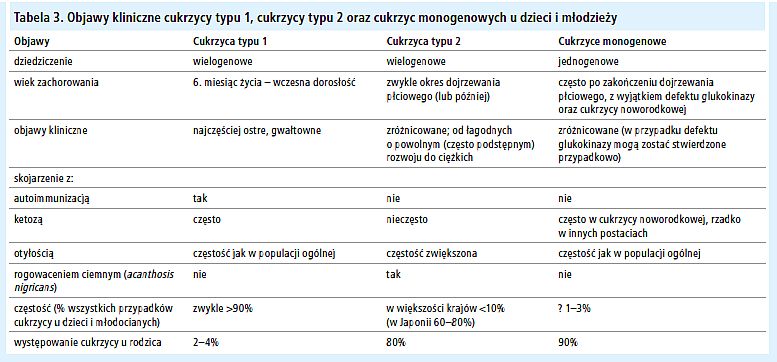
W tabeli 3 zestawiono cechy charakterystyczne cukrzycy typu 1 u dzieci i młodzieży z cechami cukrzycy typu 2 i cukrzyc monogenowych. Cukrzycę typu 2 szerzej omówiono w rozdziale "Cukrzyca typu 2 u dzieci i młodzieży".
Cukrzyce monogenowe
Defekt genetyczny funkcji komórki β lub działania insuliny dawniej określany jako cukrzyca typu MODY (maturity onset diabetes of the young) pierwotnie charakteryzowano jako zaburzenie o następujących cechach: początek przed ukończeniem 25. roku życia, autosomalny dominujący typ dziedziczenia, cukrzyca bez cukrzycowej kwasicy ketonowej.[22,57,61] Ta klasyczna definicja MODY stała się nieużyteczna z powodu występowania u dzieci cukrzycy typu 2, często spełniającej wszystkie wymienione kryteria (B, C).[6] Ponadto analiza molekularna podłoża genetycznego ujawniła, że w obrębie tych starych, szerokich kategorii istnieją znaczne różnice pomiędzy podgrupami o różnym uwarunkowaniu genetycznym, dlatego bardziej właściwe jest używanie tych podgrup genetycznych, co znalazło odzwierciedlenie w klasyfikacjach ADA i WHO (E) (tab. 2). Nasilenie hiperglikemii, zapotrzebowanie na insulinę oraz ryzyko wystąpienia późniejszych powikłań różni się znacznie w poszczególnych cukrzycach monogenowych (B)[19] (patrz rozdz. "Rozpoznawanie i leczenie cukrzyc monogenowych u dzieci i młodzieży").
Cukrzyca noworodkowa
Mianem cukrzycy noworodkowej określa się hiperglikemię wymagającą leczenia insuliną, która występuje w pierwszych trzech miesiącach życia.
Cukrzyca dziedziczona mitochondrialnie
Cukrzyca dziedziczona mitochondrialnie zwykle jest związana z głuchotą czuciowo-nerwową (odbiorczą) oraz charakteryzuje się postępującą, nieautoimmunizacyjną niewydolnością komórek β.
Mukowiscydoza i cukrzyca
Cukrzyca związana z mukowiscydozą (cystic fibrosis related diabetes – CFRD) wywołana jest głównie niedoborem insuliny, jednak insulinooporność w ostrej fazie choroby, spowodowana infekcjami oraz lekami (leki rozszerzające oskrzela, glikokortykosteroidy), może także sprzyjać upośledzeniu tolerancji glukozy i rozwojowi cukrzycy.
Cukrzyca polekowa
Hiperglikemia stresowa
Opisywano występowanie hiperglikemii stresowej u około 5% dzieci zgłaszających się na szpitalny oddział ratunkowy. Najczęściej obserwowano u nich nagłe zachorowanie lub uraz; obrażenia w przebiegu wypadku, drgawki gorączkowe lub podwyższoną temperaturę ciała (>39°C).[77]
Zalecenia
Piśmiennictwo
1. The Eurodiab Ace Study Group and The Eurodiab Ace Substudy 2 Study Group: Familial risk of type I diabetes in European children. Diabetologia, 1998; 41: 1151–1156
2. American Diabetes Association: Type 2 diabetes in children and adolescents. Pediatrics, 2000; 105: 671–680
3. Incidence and trends of childhood Type 1 diabetes worldwide 1990–1999. Diabet. Med., 2006; 23: 857–866
4. American Diabetes Association: Diagnosis and classification of diabetes mellitus. Diabetes Care, 2009; 32 suppl. 1: S62–S67
5. Al Uzri A., Stablein D.M., Cohn A.: Posttransplant diabetes mellitus in pediatric renal transplant recipients: a report of the North American Pediatric Renal Transplant Cooperative Study (NAPRTCS). Transplantation, 2001; 72: 1020–1024
6. American Diabetes Association: Type 2 diabetes in children and adolescents. American Diabetes Association. Diabetes Care, 2000; 23: 381–389
7. Barrett J.C.,Clayton D.G., Concannon P., Akolkar B., Cooper J.D., Erlich H.A., Julier C., Morahan G., Nerup J., Nierras C., Plagnol V., Pociot F., Schuilenburg H., Smyth D.J., Stevens H., Todd J.A., Walker N.M., Rich S.S.: Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet., 2009
8. Bennett C.L., Christie J., Ramsdell F., Brunkow M.E., Ferguson P.J., Whitesell L., Kelly T.E., Saulsbury F.T., Chance P.F., Ochs H.D.: Theimmune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet., 2001: 27: 20–21
9. Bhisitkul D.M., Vinik A.I., Morrow A.L., She J.X., Shults J., Powers A.C., Maclaren N.K.: Prediabetic markers in children with stress hyperglycemia. Arch. Pediatr. Adolesc. Med., 1996; 150: 936–941
10. Cucca F., Goy J.V., Kawaguchi Y., Esposito L., Merriman M.E., Wilson A.J., Cordell H.J., Bain S.C., Todd J.A.: A male-female bias in type 1 diabetes and linkage to chromosome Xp in MHC HLA-DR3-positive patients. Nat. Genet., 1998; 19: 301–302
11. Dahlquist G.G., Forsberg J., Hagenfeldt L., Boman J., Juto P.: Increased prevalence of enteroviral RNA in blood spots from newborn children who later developed type 1 diabetes: a population-based case-control study. Diabetes Care, 2004; 27: 285–286
12. Dobson L., Hattersley A.T., Tiley S., Elworthy S., Oades P.J., Sheldon C.D.: Clinical improvement in cystic fibrosis with early insulin treatment. Arch. Dis. Child., 2002; 87: 430–431
13. Dobson L., Sheldon C.D., Hattersley A.T.: Conventional measures underestimate glycemia in cystic fibrosis patients. Diabet. Med., 2004: 21: 691–696
14. Dorman J.S.,Bunker C.H.: HLA-DQ locus of the human leukocyte antigen complex and type 1 diabetes mellitus: a HuGE review. Epidemiol. Rev., 2000: 22: 218–227
15. Dorman J.S., Steenkiste A.R., O'leary L.A., Mccarthy B.J., Lorenzen T., Foley T.P.: Type 1 diabetes in offspring of parents with type 1 diabetes: the tip of an autoimmune iceberg? Pediatr. Diabetes, 2000: 1: 17–22
16. Dotta F., Censini S., Van Halteren A.G., Marselli L., Masini M., Dionisi S., Mosca F., Boggi U., Muda A.O., Prato S.D., Elliott J.F., Covacci A., Rappuoli R., Roep B.O., Marchetti P.: Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc. Natl. Acad. Sci. USA, 2007; 104: 5115–5120
17. Drachenberg C.B.,Klassen D.K.,Weir M.R.,Wiland A., Fink J.C., Bartlett S.T., Cangro C.B., Blahut S., Papadimitriou J.C.: Islet cell damage associated with tacrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation, 1999: 68: 396–402
18. Dunger D.B., Sperling M.A., Acerini C.L., Bohn D.J., Daneman D., Danne T.P., Glaser N.S., Hanas R., Hintz R.L., Levitsky L.L., Savage M.O., Tasker R.C., Wolfsdorf J.I.: European Society for Paediatric Endocrinology/Lawson Wilkins Pediatric Endocrine Society consensus statement on diabetic ketoacidosis in children and adolescents. Pediatrics, 2004; 113: e133–e140
19. Ehtisham S., Hattersley A.T., Dunger D.B., Barrett T.G.: First UK survey of paediatric type 2 diabetes and MODY. Arch. Dis. Child., 2004: 89: 526–529
20. El Hashimy M., Angelico M.C., Martin B.C., Krolewski A.S., Warram J.H.: Factors modifying the risk of IDDM in offspring of an IDDM parent. Diabetes, 1995; 44: 295–299
21. Erlich H., Valdes A.M., Noble J., Carlson J.A., Varney M., Concannon P., Mychaleckyj J.C., Todd J.A., Bonella P., Fear A.L., Lavant E., Louey A., Moonsamy P.: HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes, 2008; 57: 1084–1092
22. Fajans S.S., Bell G.I., Polonsky K.S.: Molecular mechanisms and clinical pathophysiology of maturity onset diabetes of the young. N. Engl. J. Med., 2001; 345: 971–980
23. Fourlanos S., Varney M.D., Tait B.D., Morahan G., Honeyman M.C., Colman P.G., Harrison L.C.: The rising incidence of type 1 diabetes is accounted for by cases with lower-risk human leukocyte antigen genotypes. Diabetes Care, 2008; 31: 1546–1549
24. Gale E.A., Gillespie K.M.: Diabetes and gender. Diabetologia, 2001; 44: 3–15
25. Gepts W .: Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes, 1965: 14: 619–633
26. Gillespie K.M., Bain S.C., Barnett A.H., Bingley P.J., Christie M.R.,Gill G.V., Gale E.A.: The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet, 2004; 364: 1699–1700
27. Gillespie K.M., Gale E.A., Bingley P.J.: High familial risk and genetic susceptibility in early onset childhood diabetes. Diabetes, 2002; 51: 210–214
28. Gloyn A.L., Pearson E.R., Antcliff J.F., Proks P., Bruining G.J., Slingerland A.S., Howard N., Srinivasan S., Silva J.M., Molnes J., Edghill E.L., Frayling T.M., Temple I.K., Mackay D., Shield J.P., Sumnik Z., Van Rhijn A., Wales J.K., Clark P., Gorman S., Aisenberg J., Ellard S., Njolstad P.R., Ashcroft F.M., Hattersley A.T.: Activating mutations in the gene encoding the ATP-sensitive potassium channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med., 2004; 350: 1838–1849
29. Green A., Brutti G., Patterson C.C., Dahlquist G., Soltesz G., Schober E., Weets I., Vandevalle C., Gorus F., Coeckelberghs M., Du C.M., Christov V., Tzaneva V., Iotova V., Roglic G., Vavrinec J., Olsen B.S., Svendsen A.J., Kreutzfeldt J., Lund E., Poodar T., Tuomilehto J., Karvonen M., Levymarchal C., Czernichow P.: Variation and trends in incidence of childhood diabetes in Europe. Lancet, 2000: 355: 873–876
30. Guo J.J., Keck P.E. Jr, Corey-Lisle P.K., Li H., Jiang D., Jang R., L'italien G.J.: Risk of diabetes mellitus associated with atypical antipsychotic use among patients with bipolar disorder: A retrospective, population based, case-control study. J. Clin. Psychiatry, 2006; 67: 1055–1061
31. Harjutsalo V., Podar T., Tuomilehto J.: Cumulative incidence of type 1 diabetes in 10,168 siblings of Finnish young-onset type 1 diabetic patients. Diabetes, 2005; 54: 563–569
32. Harjutsalo V., Sjoberg L., Tuomilehto J.: Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet, 2008: 371: 1777–1782
33. Harris R., Donahue K., Rathore S.S., Frame P., Woolf S.H., Lohr K.N.: Screening adults for type 2 diabetes: a review of the evidence for the U.S. Preventive Services Task Force. Ann. Intern. Med., 2003; 138: 215–229
34. Hemminki K., Li X., Sundquist J., Sundquist K.: Familial association between type 1 diabetes and other autoimmune and related diseases. Diabetologia, 2009; 52: 1820–1828
35. Hermann R., Laine A.P., Johansson C., Niederland T., Tokarska L., Dziatkowiak H., Ilonen J., Soltesz G.: Transient but not permanent neonatal diabetes mellitus is associated with paternal uniparental isodisomy of chromosome 6. Pediatrics, 2000; 105: 49–52
36. Herskowitz R.D.,Wolfsdorf J.I., Ricker A.T., Vardi P., Dib S., Soeldner J.S., Eisenbarth G.S.: Transient hyperglycemia in childhood: identification of a subgroup with imminent diabetes mellitus. Diabetes Res., 1988; 9: 161–167
37. Herskowitz-Dumont R., Wolfsdorf J.I., Jackson R.A., Eisenbarth G.S.: Distinction between transient hyperglycemia and early insulin-dependent diabetes mellitus in childhood: a prospective study of incidence and prognostic factors. J. Pediatr., 1993; 123: 347–354
38. Hoerger T.J., Harris R., Hicks K.A., Donahue K., Sorensen S., Engelgau M.: Screening for type 2 diabetes mellitus: a cost-effectiveness analysis. Ann. Intern. Med., 2004; 140: 689–699
39. Ikegami H., Fujisawa T., Kawabata Y., Noso S., Ogihara T.: Genetics of type 1 diabetes: similarities and differences between Asian and Caucasian populations. Ann. NY Acad. Sci., 2006; 1079: 51–59
40. Ilonen J., Reijonen H., Green A., Reunanen A., Knip M., Simell O., Akerblom H.K.: Geographical differences within Finland in the frequency of HLA-DQ genotypes associated with type 1 diabetes susceptibility. Eur. J. Immunogenet., 2000; 27: 225–230
41. International Diabetes Federation: Diabetes Atlas. Second. Ref Type: Serial (Book, Monograph) 2003
42. Kawasaki E., Matsuura N., Eguchi K.: Type 1 diabetes in Japan. Diabetologia, 2006; 49: 828–836
43. Kukko M., Virtanen S.M., Toivonen A., Simell S., Korhonen S., Ilonen J., Simel O., Knip M.: Geographical variation in risk HLA-DQB1 genotypes for type 1 diabetes and signs of beta-cell autoimmunity in a high-incidence country. Diabetes Care, 2004; 27: 676–681
44. Kulmala P., Savola K., Reijonen H., Veijola R., Vahasalo P., Karjalainen J., Tuomilehto-W olf E., Ilonen J., Tuomilehto J., Akerblom H.K., Knip M.: Genetic markers, humoral autoimmunity, and prediction of type 1 diabetes in siblings of affected children. Childhood Diabetes in Finland Study Group. Diabetes, 2000; 49: 48–58
45. Lambert A.P., Gillespie K.M., Thomson G., Cordell H.J., Todd J.A., Gale E.A., Bingley P.J.: Absolute risk of childhood-onset type 1 diabetes defined by human leukocyte antigen class II genotype: a population-based study in the United Kingdom. J. Clin. Endocrinol. Metab., 2004; 89: 4037–4043
46. Levy-Marchal C., Patterson C., Green A.: Variation by age group and seasonality at diagnosis of childhood IDDM in Europe. The EURODIAB ACE Study Group. Diabetologia, 1995; 38: 823–830
47. Levy-Marchal C., Patterson C.C., Green A.: Geographical variation of presentation at diagnosis of type I diabetes in children: the EURODIAB study. European and Dibetes. Diabetologia, 2001: 44 (suppl. 3): B75–B80
48. Lonnrot M., Salminen K., Knip M., Savola K., Kulmala P., Leinikki P., Hyypia T., Akerblom H.K., Hyoty H.: Enterovirus RNA in serum is a risk factor for beta-cell autoimmunity and clinical type 1 diabetes: a prospective study. Childhood Diabetes in Finland (DiMe) Study Group. J. Med. Virol., 2000: 61: 214–220
49. Lorenzen T., Pociot F., Hougaard P., Nerup J.: Longterm risk of IDDM in first-degree relatives of patients with IDDM. Diabetologia, 1994; 37: 321–327
50. Lorenzen T., Pociot F., Stilgren L., Kristiansen O.P., Johannesen J., Olsen P.B., Walmar A., Larsen A., Albrechtsen N.C., Eskildsen P.C., Andersen O.O., Nerup J.: Predictors of IDDM recurrence risk in offspring of Danish IDDM patients. Danish IDDM Epidemiology and Genetics Group. Diabetologia, 1998; 41: 666–673
51. Maclaren N., Lan M., Coutant R., Schatz D., Silverstein J., Muir A., Clare-Salzer M., She J.X., Malone J., Crockett S., Schwartz S., Quattrin T., Desilva M., Vander V.P., Notkins A., Krischer J.: Only multiple autoantibodies to islet cells (ICA), insulin, GAD65, IA-2 and IA-2beta predict immune-mediated (Type 1) diabetes in relatives. J. Autoimmun., 1999; 12: 279–287
52. Maes B.D., Kuypers D., Messiaen T., Evenepoel P., Mathieu C., Coosemans W ., Pirenne J., Vanrenterghem Y.F.: Posttransplantation diabetes mellitus in FK-506-treated renal transplant recipients: analysis of incidence and risk factors. Transplantation, 2001; 72: 1655–1661
53. Massa O., Iafusco D., D'amato E., Gloyn A.L., Hattersley A.T., Pasquino B., Tonini G., Dammacco F., Zanette G., Meschi F., Porzio O., Bottazzo G., Crino A., Lorini R., Cerutti F., Vanelli M., Barbetti F.: KCNJ11 activating mutations in Italian patients with permanent neonatal diabetes. Hum. Mutat., 2005; 25: 22–27
54. Metz C., Cave H., Bertrand A.M., Deffert C., Gueguen-Giroux B., Czernichow P., Polak M.: Neonatal diabetes mellitus: chromosomal analysis in transient and permanent cases. J. Pediatr., 2002; 141: 483–489
55. Moran A., Dunitz J., Nathan B., Saeed A., Holme B., Thomas W .: Cystic fibrosis-related diabetes: current trends in prevalence, incidence and mortality. Diabetes Care, 2009; 32: 1626–1631
56. Moran A., Hardin D., Rodman D., Allen H.F., Beall R.J., Borowitz D., Brunzell C., Campbell P.W. Iii, Chesrown S.E., Duchow C., Fink R.J., Fitzsimmons S.C., Hamilton N., Hirsch I., Howenstine M.S., Klein D.J., Madhun Z., Pencharz P.B., Quittner A.L., Robbins M.K., Schindler T., Schissel K., Schwarzenberg S.J., Stallings V.A., Zipf W.B.: Diagnosis, screening and management of cystic fibrosis related diabetes mellitus: a consensus conference report. Diabetes Res. Clin. Pract., 2009; 45: 61–73
57. Murphy R., Ellard S., Hattersley A.T.: Clinical implications of a molecular genetic classification of monogenic ß–cell diabetes. Nat. Clin. Pract. Endocrinol. Metab., 2008; 4: 200
58. Njolstad P.R., Sovik O., Cuesta-Munoz A., Bjorkhaug L., Massa O., Barbetti F., Undlien D.E., Shiota C., Magnuson M.A., Molven A., Matschinsky F.M., Bell G.I.: Neonatal diabetes mellitus due to complete glucokinase deficiency. N. Engl. J. Med., 2001; 344: 1588–1592
59. Nousia-Arvanitakis S., Galli-Tsinopoulou A., Karamouzis M.: Insulin improves clinical status of patients with cystic-fibrosis-related diabetes mellitus. Acta paediatr., 2001; 90: 515–519
60. OLmos P., A'hern R., Heaton D.A., Millward B.A., Risley D., Pyke D.A., Leslie R.D.: The significance of the concordance rate for type 1 (insulin-dependent) diabetes in identical twins. Diabetologia, 1988; 31: 747–750
61. Owen K., Hattersley A.T.: Maturity-onset diabetes of the young: from clinical description to molecular genetic characterization. Best Pract. Res. Clin. Endocrinol. Metab., 2001; 15: 309–323
62. Patterson C.C., Dahlquist G.G., Gyurus E., Green A., Soltesz G.: Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: a multicentre prospective registration study. Lancet, 2009; 373: 2027–2033
63. Pinhas-Hamiel O., Zeitler P.: The global spread of type 2 diabetes mellitus in children and adolescents. J. Pediatr., 2005; 146: 693–700
64. Pui C.H., Burghen G.A., Bowman W.P., Aur R.J.: Risk factors for hyperglycemia in children with leukemia receiving L-asparaginase and prednisone. J. Pediatr., 1981; 99: 46–50
65. Rasilainen S.,Ylipaasto P., Roivainen M., Bouwens L., Lapatto R., Hovi T., Otonkoski T.: Mechanisms of beta cell death during restricted and unrestricted enterovirus infection. J. Med. Virol., 2004; 72: 451–461
66. Reardon W ., Ross R.J., Sweeney M.G., Luxon L.M., Pembrey M.E., Harding A.E., Trembath R.C.: Diabetes mellitus associated with a pathogenic point mutation in mitochondrial DNA. Lancet, 1992; 340: 1376–1379
67. Richardson S.J., Willcox A., Bone A.J., Foulis A.K., Morgan N.G.: The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia, 2009; 52: 1143–1151
68. Sabbah E., Savola K., Ebeling T., Kulmala P., Vahasalo P., Ilonen J., Salmela P.I., Knip M.: Genetic, autoimmune, and clinical characteristics of childhoodand adult-onset type 1 diabetes. Diabetes Care, 2000; 23: 1326–1332
69. Sadeharju K., Hamalainen A.M., Knip M., Lonnrot M., Koskela P., Virtanen S.M., Ilonen J., Akerblom H.K., Hyoty H.: Enterovirus infections as a risk factor for type I diabetes: virus analyses in a dietary intervention trial. Clin. Exp. Immunol., 2003: 132: 271–277
70. Schatz D.A.,Kowa H.,Winter W .E., Riley W .J.: Natural history of incidental hyperglycemia and glycosuria of childhood. J. Pediatr., 1989; 115: 676–680
71. Shehadeh N., On A., Kessel I., Perlman R., Even L., Naveh T., Soloveichik L., Etzioni A.: Stress hyperglycemia and the risk for the development of type 1 diabetes. J. Pediatr. Endocrinol. Metab., 1997; 10: 283–286
72. Silverstein J., Klingensmith G., Copeland K., Plotnick L., Kaufman F., Laffel L., Deeb L., Grey M., Anderson B., Holzmeister L.A., Clark N.: Care of children and adolescents with type 1 diabetes: a statement of the American Diabetes Association. Diabetes Care, 2005; 28: 186–212
73. Skyler J.S., Krischer J.P., Wolfsdorf J., Cowie C., Palmer J.P., Greenbaum C., Cuthbertson D., Rafkin-Mervis L.E., Chase H.P., Leschek E.: Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial – Type 1. Diabetes Care, 2005; 28: 1068–1076
74. Steck A.K., Barriga K.J., Emery L.M., Fialloscharer R.V., Gottlieb P.A., Rewers M.J.: Secondary attack rate of type 1 diabetes in Colorado families. Diabetes Care, 2005; 28: 296–300
75. Thunander M., Petersson C., Jonzon K., Fornander J., Ossiansson B., Torn C., Edvardsson S., Landin-Olsson M.: Incidence of type 1 and type 2 diabetes in adults and children in Kronoberg, Sweden. Diabetes Res. Clin. Pract., 2008; 82: 247–255
76. Urakami T., Suzuki J., Yoshida A., Saito H., Mugishima H.: Incidence of children with slowly progressive form of type 1 diabetes detected by the urine glucose screening at schools in the Tokyo Metropolitan Area. Diabetes Res. Clin. Pract., 2008; 80: 473–476
77. Valerio G., Franzese A., Carlin E., Pecile P., Perini R., Tenore A.: High prevalence of stress hyperglycemia in children with febrile seizures and traumatic injuries. Acta Paediatr., 2001; 90: 618–622
78. Van Den Ouweland J.M., Lemkes H.H., Ruitenbeek W ., Sandkuijl L.A., De Vijlder M.F., Struyvenberg P.A., Van De Kamp J.J., Maassen J.A.: Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet., 1992; 1: 368–371
79. Vandewalle C.L., Coeckelberghs M.I., De L., Du I.C.M., Schuit F.C., Pipeleers D.G., Gorus F.K.: Epidemiology, clinical aspects, and biology of IDDM patients under age 40 years. Comparison of data from Antwerp with complete ascertainment with data from Belgium with 40% ascertainment. The Belgian Diabetes Registry. Diabetes Care, 1997; 20: 1556–1561
80. Vardi P., Shehade N., Etzioni A., Herskovits T., Soloveizik L., Shmuel Z., Golan D., Barzilai D., Benderly A.: Stress hyperglycemia in childhood: a very high risk group for the development of type I diabetes. J. Pediatr., 1990; 117: 75–77
81. Verge C.F., Gianani R., Kawasaki E., Yu L., Pietropaolo M., Jackson R.A., Chase H.P., Eisenbarth G.S.: Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes, 1996; 45: 926–933
82. Verge C.F., Stenger D., Bonifacio E., Colman P.G., Pilcher C., Bingley P.J., Eisenbarth G.S.: Combined use of autoantibodies (IA-2 autoantibody, GAD autoantibody, insulin autoantibody, cytoplasmic islet cell antibodies) in type 1 diabetes: Combinatorial Islet Autoantibody Workshop. Diabetes, 1998; 47: 1857–1866
83. Von Muhlendahl K.E., Herkenhoff H.: Long-term course of neonatal diabetes. N. Engl. J. Med., 1995; 333: 704–708
84. Warram J.H., Krolewski A.S., Gottlieb M.S., Kahn C.R.: Differences in risk of insulin-dependent diabetes in offspring of diabetic mothers and diabetic fathers. N. Engl. J. Med., 1984; 311: 149–152
85. Weets I., Kaufman L., Van Der A.B., Crenier L., Rooman R.P., De Block C., Casteels K., Weber E., Coeckelberghs M., Laron Z., Pipeleers D.G., Gorus F.K.: Seasonality in clinical onset of type 1 diabetes in Belgian patients above the age of 10 is restricted to HLA-DQ2/DQ8-negativemales,which explains themale to female excess in incidence. Diabetologia, 2004; 47: 614–621
86. World Health Organisation: Definition, Diagnosis and Classification of Diabetes Mellitus and its Complications. Part 1: Diagnosis and Classification of Diabetes Mellitus. WHO/NCD/NCS/99.2. Geneva. Ref Type: Report. 1999
87. Yankaskas J.R., Marshall B.C., Sufian B., Simon R.H., Rodman D.: Cystic fibrosis adult care: consensus conference report. Chest, 2004; 125: S1–S39
88. Yoon J.W., Austin M., Onodera T., Notkins A.L.: Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N. Engl. J. Med., 1979; 300: 1173–1179
Konflikt interesów
Autorzy nie zgłosili konfliktu interesów.