
Tłumaczyła lek. Iwona Rywczak
Konsultowała prof. dr hab. n. med. Przemysława Jarosz-Chobot, Klinika Diabetologii Dziecięcej
Śląskiego Uniwersytetu Medycznego w Katowicach
Skróty: CFRD – cukrzyca związana z mukowiscydozą, GAD – dekarboksylaza kwasu glutaminowego, GKS – glik
Podsumowanie i zalecenia
- Kryteria rozpoznawania cukrzycy opierają się na labolatoryjnym oznaczeniu stężenia glukozy w osoczu oraz występowaniu objawów klinicznych lub ich braku (E). W procesie diagnostycznym cukrzycy nie należy wykorzystywać pomiaru stężenia glukozy glukometrem we krwi pobranej z opuszki palca (E).
- Znacznie zwiększone stężenie glukozy we krwi potwierdza rozpoznanie cukrzycy. Jeżeli we krwi lub moczu stwierdza się obecność związków ketonowych, konieczne jest natychmiastowe wdrożenie leczenia i skierowanie dziecka do szpitala w tym samym dniu, aby uniknąć rozwoju kwasicy ketonowej (A).
- Rozpoznania cukrzycy nie należy ustalać na podstawie jednego oznaczenia stężenia glukozy w osoczu. W niejasnych przypadkach ustalenie rozpoznania może wymagać dłuższej obserwacji, z oznaczeniem glikemii na czczo i/lub 2 h po posiłku i/lub wykonania doustnego testu tolerancji glukozy (OGTT) (E). Wykonanie OGTT nie jest jednak konieczne i nie należy go przeprowadzać, jeżeli rozpoznanie cukrzycy można ustalić na podstawie pomiaru glikemii na czczo, przygodnej lub poposiłkowej, ponieważ może to doprowadzić do hiperglikemii (E).
- Hiperglikemia wykryta w okresie działania czynnika stresowego dla organizmu (np. ostrego zakażenia, urazu, zabiegu chirurgicznego, zaburzeń w układzie krążenia, oddechowym lub innego) może przemijać. Choć wymaga leczenia, w postaci izolowanej nie jest kryterium rozpoznania cukrzycy (E).
- W diagnostyce różnicowej należy rozważyć inne typy
cukrzycy, jeżeli u dziecka nie stwierdzono autoprzeciwciał
związanych z cukrzycą, a także (B):
· w wywiadzie rodzinnym stwierdza się cukrzycę o autosomalnie
dominującym typie dziedziczenia
· cukrzycę rozpoznano w pierwszych 6 miesiącach
życia
· stwierdza się łagodną hiperglikemię na czczo (5,5–
8,5 mmol [100–150 mg/dl]), która nie postępuje
(dotyczy to zwłaszcza młodych, nieotyłych osób,
bez objawów cukrzycy)
· współwystepują dodatkowe zaburzenia, takie jak
głuchota, zanik nerwu wzrokowego, objawy innych
znanych zespołów chorobowych
· wywiad potwierdza ekspozycję na leki toksyczne
dla komórek ß lub powodujące insulinooporność.
-Rozróżnienie pomiędzy cukrzycą typu 1, typu 2, monogenową i jej innymi typami ma istotne znaczenie dla
leczenia i edukacji pacjenta (E). W niejasnych przypadkach w rozpoznaniu typu cukrzycy pomocne mogą
być takie badania, jak:
· autoprzeciwciała związane z cukrzycą: przeciwciała
przeciwko izoformie dekarboksylazy kwasu glutaminowego o masie 65 kDA (GAD), przeciwciała przeciwko
związanym z fosfatazą tyrozynową cząsteczkom
IA2, autoprzeciwciała przeciwinsulinowe (IAA) oraz
przeciwciała przeciwko transporterowi cynku swoistemu
dla komórek ß (ZnT8) – obecność co najmniej
jednego rodzaju z wymienionych autoprzeciwciał
potwierdza rozpoznanie cukrzycy typu 1 (A)
· OGTT (A)
· hemoglobina A1c (HbA1c) (B).
- Badania molekularne mogą być pomocne w ustaleniu rozpoznania i wyborze metody leczenia u dzieci z podejrzeniem cukrzycy monogenowej. U wszystkich chorych, u których cukrzycę rozpoznano w pierwszych 6 miesiącach życia, należy niezwłocznie przeprowadzić molekularne badania genetyczne w celu określenia podtypu cukrzycy noworodków (neonatal diabetes mellitus – NDM), ponieważ w tej grupie wiekowej cukrzyca typu 1 występuje wyjątkowo rzadko (B). U osób w wieku >6 miesięcy badania genetyczne należy wykonać w przypadku niewykrycia autoprzeciwciał (zwłaszcza gdy autoprzeciwciała oznaczono w momencie rozpoznania) oraz obecności objawów klinicznych sugerujących cukrzycę monogenową (E).
Definicja
Mianem „cukrzycy” określa się grupę chorób metabolicznych
charakteryzujących się przewlekłą
hiperglikemią wynikającą z zaburzenia wydzielania
i/lub działania insuliny. Niedostateczne wydzielanie
insuliny i/lub zmniejszona odpowiedź
tkanek na insulinę upośledza złożone działanie
insuliny w tkankach docelowych, co skutkuje zaburzeniem
metabolizmu węglowodanów, lipidów i białek. U chorego może być obecne zarówno upośledzenie
wydzielania, jak i funkcji insuliny.1,2
Etiologia cukrzycy jest wieloczynnikowa, jednak
większość przypadków cukrzycy można
zakwalifikować do dwóch szerokich kategorii
etiopatogenetycznych (p. niżej): cukrzycy typu 1, w której występuje całkowity brak wydzielania insuliny,
lub cukrzycy typu 2, charakteryzującej się
opornością na insulinę oraz niedostatecznym wyrównawczym
wydzielaniem insuliny. Podczas gdy
cukrzyca typu 1 jest najczęstszą postacią choroby u młodych osób w wielu populacjach, szczególnie
rasy białej, cukrzyca typu 2 jest coraz większym
problemem zdrowia publicznego na całym świecie
(p. rozdz. Cukrzyca typu 2 u dzieci i młodzieży).3
Kryteria diagnostyczne cukrzycy u dzieci i młodzieży
Kryteria diagnostyczne cukrzycy opierają się
na ocenie stężenia glukozy we krwi oraz występowaniu
objawów klinicznych lub ich braku.1,4 W procesie diagnostycznym cukrzycy stosuje się
różne metody (tab. 1.), a w przypadku wątpliwej
hiperglikemii konieczne jest powtórzenie badań w celu potwierdzenia rozpoznania.
• Cukrzyca u młodych osób zwykle rozpoczyna
się od wystąpienia typowych objawów, takich
jak wielomocz, zwiększone pragnienie, mimowolne
oddawanie moczu, oddawanie moczu w nocy (nocturia) oraz zmniejszenie masy ciała. U niektórych pacjentów obecne jest także
wzmożone łaknienie i zaburzenia widzenia
(zamazane widzenie). Przewlekłej hiperglikemii
może towarzyszyć upośledzenie wzrastania i większa podatność na niektóre zakażenia.
• W najcięższej postaci może się rozwinąć cukrzycowa
kwasica ketonowa lub rzadziej nieketonowy
zespół hiperosmolalny, które mogą doprowadzić
do stuporu lub śpiączki, a w przypadku
braku skutecznego leczenia – do zgonu.
• U pacjentów, u których stwierdza się wymienione
objawy, prostym i czułym badaniem przesiewowym
jest ocena glukozurii i ketonurii za
pomocą testu paskowego lub pomiar stężenia
glukozy i związków ketonowych za pomocą glukometru.
Jeżeli stężenie glukozy we krwi jest
zwiększone, dziecko należy niezwłocznie skierować
do ośrodka wyspecjalizowanego w leczeniu
cukrzycy u dzieci. Potwierdzenie hiperglikemii w kolejnym dniu jest zbędne, a w przypadku
obecności związków ketonowych we krwi konieczne
jest zawsze szybkie wdrożenie leczenia
ze względu na ryzyko nagłego rozwoju kwasicy
ketonowej.
• W celu potwierdzenia rozpoznania wymagane
jest oficjalne labolatoryjne oznaczenie stężenia
glukozy w osoczu metodą z oksydazą glukozy, a nie przesiewowe oznaczenie glikemii we krwi
włośniczkowej glukometrem.
• Rozpoznanie cukrzycy może być wątpliwe w przypadku:
– braku objawów klinicznych (np. w przypadkowo
wykrytej hiperglikemii lub u dzieci uczestniczących w badaniach przesiewowych)
– stwierdzenia łagodnych lub nietypowych objawów
cukrzycy
– hiperglikemii w przebiegu ostrego zakażenia,
po urazie, w przebiegu zaburzeń układu krążenia
lub wywołanej innym czynnikiem stresowym,
ponieważ w takich sytuacjach może
przemijać i nie należy jej traktować jako potwierdzenie
rozpoznania cukrzycy. W wymienionych przypadkach cukrzycy nie
należy rozpoznawać na podstawie jednokrotnego
oznaczenia stężenia glukozy w osoczu. Ustalenie
rozpoznania może wymagać dłuższej obserwacji z oznaczeniem glikemii na czczo oraz 2 h po posiłku
i/lub wykonania OGTT.
• Wykonanie OGTT nie jest konieczne i nie należy
go przeprowadzać, jeżeli rozpoznanie cukrzycy
można ustalić na podstawie pomiaru glikemii
na czczo, przygodnej lub poposiłkowej, ponieważ
może to doprowadzić do hiperglikemii. OGTT
jest rzadko wskazany w diagnostyce cukrzycy
typu 1 u dzieci i młodzieży, lecz może on być
pomocny w rozpoznawaniu innych postaci choroby,
takich jak cukrzyca typu 2, cukrzyca monogenowa
lub związana z mukowiscydozą (cystic
fibrosis related diabetes – CFRD). W razie
wątpliwości należy okresowo powtarzać OGTT
do czasu ustalenia jednoznacznego rozpoznania.
• W diagnostyce cukrzycy można wykorzystać
oznaczenie HbA1c. Warunkiem jest zastosowanie
testów poddanych rygorystycznej procedurze
zachowania jakości, standaryzowanych
do międzynarodowych wartości referencyjnych
oraz brak okoliczności uniemożliwiających
dokładny pomiar stężenia HbA1c.2,5 Jednak w szybko rozwijającej się cukrzycy, na przykład u niektórych dzieci chorych na cukrzycę typu 1,
HbA1c może być tylko nieznacznie zwiększone
mimo obecności typowych objawów choroby.
| Tabela 1. Kryteria diagnostyczne cukrzycy1,2 |
|---|
| • Typowe objawy cukrzycy lub przełom hiperglikemiczny oraz stężenie glukozy w osoczu ≥11,1 mmol/l (200 mg/dl) lub • Glikemia na czczo ≥7,0 mmol/l (≥126 mg/dl); oznaczenie należy wykonać co najmniej po 8 ha na czczo lub • Glikemia 2 h po obciążeniu w trakcie OGTT ≥11,1 mmol/l (≥200 mg/dl)a Test należy przeprowadzić z obciążeniem odpowiadającym 75 g bezwodnej glukozyrozpuszczonej w wodzie lub 1,75 g/kg mc. (maks. 75 g) lub • HbA1c >6,5%bOznaczenie należy wykonać w laboratorium stosującym metodę z certyfikatem NGSP i wystandaryzowaną do metody wykorzystanej w badaniu DCCT. |
| a Jeśli hiperglikemia jest wątpliwa, należy powtórzyć badanie w celu potwierdzenia rozpoznania na podstawie tych kryteriów.
b Wartość HbA1c <6,5% nie wyklucza cukrzycy rozpoznanej na podstawie pomiarów glikemii. Rola oznaczania samego stężenia HbA1c w rozpoznaniu cukrzycy typu 1 u dzieci jest nieznana. DCCT – Diabetes Control and Complications Trial, HbA1c – hemoglobina A1c (glikowana), NGSP – National Glycohemoglobin Standardization Program, OGTT – doustny test tolerancjiglukozy |
Upośledzona tolerancja glukozy i nieprawidłowa glikemia na czczo
Upośledzona tolerancja glukozy (impaired glucose
tolerance – IGT) i nieprawidłowa glikemia na czczo
(impaired fasting glucose – IFG) są pośrednimi
stanami w naturalnym rozwoju zaburzeń metabolizmu
węglowodanów pomiędzy prawidłową homeostazą
glukozy a cukrzycą.2 Pojęć „IFG” i „IGT”
nie można używać zamiennie, ponieważ opisują
one różne nieprawidłowości w regulacji stężenia
glukozy lub różne etapy progresji zaburzeń tego
procesu. IFG jest miarą zaburzeń metabolizmu
węglowodanów w stanie podstawowym, natomiast
IGT jest dynamicznym wskaźnikiem nietolerancji
węglowodanów po obciążeniu standardową porcją
glukozy. IFG i IGT nie są jednostkami chorobowymi, u pacjentów z IFG i/lub IGT rozpoznaje się stan
przedcukrzycowy, co wskazuje na występowanie u nich względnie dużego ryzyka rozwoju cukrzycy i chorób układu krążenia.
IFG i IGT mogą być związane z zespołem metabolicznym, w którym występuje otyłość (zwłaszcza
otyłość brzuszna lub trzewna), dyslipidemia
(duże stężenie triglicerydów i/lub małe stężenie
lipoprotein o dużej gęstości [HDL]) i nadciśnienie
tętnicze. IFG i IGT mogą występować jako stany
pośrednie we wszystkich chorobach wymienionych w tabeli 2. (etiologiczna klasyfikacja cukrzycy).
U osób spełniających kryteria IGT lub IFG stężenie
glukozy na co dzień może być prawidłowe,
na co wskazują prawidłowe wartości HbA1c lub
bliskie normy, a w przypadku IGT hiperglikemię
można wykazać tylko za pomocą OGTT.
Kategorie stężenia glukozy w osoczu na czczo
(fasting plasma glucose – FPG) definiuje się w następujący
sposób:
• FPG <5,6 mmol/l (100 mg/dl) – prawidłowa glikemia
na czczo
• FPG 5,6–6,9 mmol/l (100–125 mg/dl) – nieprawidłowa
glikemia na czczo (IFG)
• FPG ≥7,0 mmol/l (126 mg/dl) – wstępne rozpoznanie
cukrzycy (konieczne potwierdzenie
rozpoznania – p. tab. 1.).
Odpowiednie kategorie w przypadku wykonania
OGTT są następujące:
• 2 h po obciążeniu glukozą <7,8 mmol/l
(140 mg/dl) – prawidłowa tolerancja glukozy
• 2 h po obciążeniu glukozą od 7,8 do <11,1 mmol/l
(140–200 mg/dl) – nieprawidłowa tolerancja
glukozy (IGT)
• 2 h po obciążeniu glukozą ≥11,1 mmol/l
(200 mg/dl) – wstępne rozpoznanie cukrzycy
(konieczne potwierdzenie rozpoznania, jak opisano
powyżej).
| Tabela 2. Etiologiczna klasyfikacja cukrzycy |
|---|
| typ 1 |
| zniszczenie komórek ß, zwykle prowadzące do całkowitego niedoboru insuliny
związane z autoimmunizacją idiopatyczne |
| typ 2 |
| może obejmować zaburzenia od dominującej insulinooporności ze względnym niedoborem insuliny do dominującego upośledzenia wydzielania z insulinoopornością lub bez niej |
| inne określone typy |
| • defekty genetyczne czynności komórek ß |
| chromosom 12, HNF1A (MODY3)
chromosom 7, GCK (MODY2) chromosom 20, HNF4B (MODY1) inne rzadkie postacie MODY: chromosom 13, IPF-1 (MODY4) chromosom 17, HNF1B (MODY5) chromosom 2, NEUROD1 (MODY6) chromosom 2, KLF11 (MODY7) chromosom 9, CEL (MODY8) chromosom 7, PAX4 (MODY9) TNDM (najczęściej związana z imprintingiem genów PLAGL1/HYMAI na chromosomie 6q24) PNDM (najczęściej związana z mutacją genu KCNJ11 kodującego Kir6.2, podjednostkę kanału potasowego zależnego od ATP komórek ß) mutacje DNA mitochondrialnego inne |
| • defekty genetyczne działania insuliny |
| insulinooporność typu A leprechaunizm zespół Rabsona i Mendenhalla cukrzyca lipoatroficzna inne |
| • choroby części zewnątrzwydzielniczej trzustki |
| zapalenie trzustki
uraz lub stan po wycięciu trzustki nowotwór mukowiscydoza hemochromatoza pankreatopatia włóknisto-wapniejąca inne |
| • endokrynopatie |
| akromegalia
zespół Cushinga glucagonoma guz chromochłonny nadczynność tarczycy somatostatinoma aldosteronoma inne |
| • wywołana lekami lub substancjami chemicznymi |
| Vacora
pentamidyna kwas nikotynowy glikokrtykosteroidy hormony tarczycy diazoksyd agoniści receptorów ß-adrenergicznych tiazydy dylantynab interferon α inne |
| • zakażenia |
| różyczka wrodzona
cytomegalia zakażenie enterowirusami inne |
| • rzadkie typy cukrzycy wywołane procesem immunologicznym |
| zespół uogólnionej sztywności („stiff-man” syndrome)
przeciwciała przeciwko receptorowi insuliny autoimmunizacyjny zespół niedoczynności wielogruczołowej typu I i II IPEX inne |
| • inne zespoły genetyczne niekiedy związane z cukrzycą |
| zespół Downa
zespół Klinefeltera zespół Turnera zespół Wolframa ataksja Friedreicha pląsawica Huntingtona zespół Laurence’a, Moona i Biedla dystrofia miotoniczna porfiria zespół Pradera i Williego inne |
| cukrzyca ciążowa (GDM) |
| Każda postać cukrzycy może, ale nie musi wymagać leczeniainsuliną na różnych etapach choroby. Stosowanie insuliny niestanowi kryterium klasyfikacji cukrzycy.
a środek do deratyzacji zawierający N-3 pirydylometylo-N’-p-nitrofenylomocznik – przyp. red. b fenytoina – przyp. red. CEL – lipaza estrów karboksylowych, HNF – czynnik jądrowyhepatocytów, IPEX – zespół dysregulacji immunologicznejz poliendokrynopatią i enteropatią sprzężonyz chromosomem X, IPF – czynnik promotora insuliny, KLF11 – zynnik 11 podobny do czynnika Kruppel, MODY – maturity-onset diabetes of the young, PAX4 – genPAX4 |
Klasyfikacja cukrzycy i inne kategorie zaburzeń regulacji stężenia glukozy
Rozpoznając cukrzycę u młodego człowieka, typ choroby
zwykle określa się na podstawie objawów klinicznych,
jednak coraz częściej możliwość ustalenia
rozpoznania klinicznego ograniczają pewne czynniki,
do których należy coraz częstsze występowanie nadwagi u młodych osób chorych na cukrzycę typu 16,7
oraz obecność cukrzycowej kwasicy ketonowej u niektórych
młodych chorych w momencie rozpoznania
cukrzycy typu 2.8,9 Poza tym wystąpienie w wieku
młodzieńczym rodzinnej postaci cukrzycy o łagodnym
przebiegu powinno nasuwać podejrzenie cukrzycy
monogenowej, która stanowi 1–4% przypadków
cukrzycy wieku rozwojowego.10-13
Klasyfikację cukrzycy w zależności od jej etiologii,
opracowaną na podstawie klasyfikacji American
Diabetes Association, przedstawiono w tabeli 2.2 Niektóre postacie choroby, takie jak cukrzyca
wywołana niektórymi lekami, zaburzeniami hormonalnymi
lub toksynami, rzadko obserwuje się u młodych osób. W Afryce i Azji Południowej nietypowe
postacie cukrzycy mogą wystąpić u starszych
dzieci, młodzieży i młodych dorosłych, a należą
do nich: nietypowa cukrzyca ze skłonnością
do ketozy, cukrzyca związana z niedożywieniem
oraz pankreopatia włóknisto-wapniejąca.14,15
Odróżnienie cukrzycy typu 1, typu 2, monogenowej i jej innych typów ma istotne znaczenie dla
leczenia i edukacji pacjenta. Poniżej wymieniono
badania, które mogą być pomocne w ustaleniu
typu cukrzycy.
• Autoprzeciwciała związane z cukrzycą: obecność
przeciwciał GAD, IA2, IAA i/lub ZnT8
potwierdza rozpoznanie cukrzycy typu 1, ponieważ
jedne z nich (a zwykle więcej) stwierdza się u 85–90% osób, u których na początku wykryto
hiperglikemię na czczo.16
• Zwiększone stężenie peptydu C na czczo pomaga
odróżnić u młodych osób nieautoimmunizacyjną
cukrzycę typu 2 przebiegającą z insulinoopornością
od cukrzycy typu 1.17 Jednak w pierwszym
roku od rozpoznania poziomy insuliny lub peptydu
C stwierdzane w cukrzycy typu 1 i 2 w dużym
stopniu pokrywają się, dlatego stężenia peptydu
C nie należy oznaczać w ostrej fazie choroby. U chorych leczonych insuliną ocena stężenia
peptydu C (test stymulacji glukagonem lub standaryzowanym
posiłkiem) przy stężeniu glukozy
wystarczająco dużym (>8 mmol/l), aby stymulować
wydzielanie peptydu C, pozwala ustalić, czy
zachowane jest jeszcze endogenne wydzielanie
insuliny. U dzieci chorych na cukrzycę typu 1 rzadko obserwuje się wydzielanie endogennej
insuliny poza okresem remisji (2–3 lata).
Inny typ cukrzycy należy rozważyć u dziecka, u którego nie wykryto autoprzeciwciał, a także:
• w wywiadzie rodzinnym stwierdza się cukrzycę o autosomalnie dominującym typie dziedziczenia
• cukrzycę rozpoznano w pierwszych 6 miesiącach
życia
• stwierdza się łagodną hiperglikemię na czczo
(5,5–8,5 mmol [100–150 mg/dl]), która nie postępuje
(dotyczy to zwłaszcza młodych, nieotyłych
osób, bez objawów cukrzycy)
• stwierdza się dodatkowe zaburzenia, na przykład
głuchotę, zanik nerwu wzrokowego, cechy
znanych zespołów chorobowych
• wywiad potwierdza narażenie na leki toksyczne
dla komórek ß lub wywołujące insulinooporność. W tabeli 3. zestawiono charakterystyczne cechy
cukrzycy typu 1 u dzieci i młodzieży z cechami cukrzycy
typu 2 i cukrzyc monogenowych. Cukrzycę
typu 2 szerzej omówiono w wytycznych ISPAD
dotyczących cukrzycy typu 23 i cukrzyc monogenowych.
18 Jednak niezależnie od typu cukrzycy, u dzieci z ciężką hiperglikemią, ketonemią i zaburzeniami
metabolicznymi w pierwszej kolejności
należy rozpocząć leczenie insuliną w celu wyrównania
tych zaburzeń.
Patogeneza cukrzycy typu 1
Cukrzyca typu 1 charakteryzuje się przewlekłym
procesem immunologicznym niszczącym komórki
ß trzustki, co prowadzi do częściowego, a zwykle
całkowitego niedoboru insuliny. W większości
przypadków (typ 1A) zniszczenie komórek ß
zachodzi na drodze autoimmunizacyjnej, tempo i nasilenie tego procesu są różne, a objawy kliniczne
występują po zniszczeniu około 90% komórek
ß. Etiologia jest wieloczynnikowa, jednak
nie wiadomo, jaką konkretnie rolę w patogenezie
cukrzycy typu 1 odgrywa predyspozycja genetyczna,
czynniki środowiskowe, układ immunologiczny i komórki ß.
Wśród autoprzeciwciał związanych z cukrzycą,
będących wskaźnikami serologicznymi procesu
autoimmunizacyjnego, wymienia się GAD, IA2,
IAA i ZnT8.16 Wytwarzanie tych przeciwciał zależy
od wieku – IAA i ZnT8 częściej stwierdza się u dzieci do 10. roku życia, natomiast GAD i IA-2
są związane ze starszym wiekiem, a GAD z płcią
żeńską.19
Predyspozycja do rozwoju cukrzycy typu 1 z autoimmunizacji jest uwarunkowana wieloma
genami, w genomowych analizach asocjacyjnych
zidentyfikowano ponad 60 loci związanych z większym
ryzykiem wystąpienia choroby.20 W około
50% ryzyko zależy od genów układu HLA.21,22 W populacji rasy białej predyspozycję genetyczną
wyznaczają swoiste kombinacje alleli HLA
DR i DQ.23 Do haplotypów największego ryzyka należą: DRB1*03:01-DQA1*05:01-DQB1*02:01 i DRB1*04-DQA1*03:01-DQB1*03:02 (określane
również zgodnie z wcześniejszą klasyfikacją
serologiczną jako DR3/DR4 lub DQ2/DQ8). Haplotypy
ochronne w odniesieniu do cukrzycy
typu 1 to DRB1*15:01-DQA1*01:02-DQB1*06:02
oraz DRB1*14:01-DQA1*01:01-DQB*05:03 i DRB1*07:01-DQA1*02:01-DQB1*03:03.24 U osób
heterozygotycznych dla dwóch haplotypów HLA o największym ryzyku (DR3/4) iloraz szans dla
rozwoju procesu autoimmunizacyjnego obejmującego
wyspy trzustki i cukrzycy typu 1 wynosi
30,24 jednak objawowa choroba rozwija się u <10%
osób z predyspozycjami do cukrzycy wynikającymi z genotypu HLA.25
Osoby obarczone zwiększonym ryzykiem rozwoju
cukrzycy typu 1 można zidentyfikować na podstawie
oznaczenia autoprzeciwciał związanych z cukrzycą, wskaźników genetycznych oraz testu
dożylnego obciążenia glukozą (intravenous glucose
tolerance test – IVGTT) i/lub OGTT.26-30
Większość czynników środowiskowych (zakaźnych
i/lub chemicznych) inicjujących niszczenie
komórek ß jest nieznana, ale proces ten zwykle
rozpoczyna się wiele miesięcy lub lat przed wystąpieniem
objawów klinicznych.28,31,32 W wielu populacjach
pojawienie się autoprzeciwciał przeciwko
wyspom trzustkowym i rozwój cukrzycy typu 1 wiązano z zakażeniem enterowirusami,33,34 a enterowirusy
wykrywano w wyspach trzustkowych
chorych na cukrzycę.35-37
Jeżeli objawy kliniczne są typowe dla cukrzycy
typu 1, ale nie stwierdza się przeciwciał, cukrzycę
klasyfikuje się jako typ 1B (idiopatyczny). W większości
przypadków dotyczy to osób pochodzenia
afrykańskiego lub azjatyckiego, jakkolwiek należy
wziąć pod uwagę możliwość występowania innych
typów choroby, na przykład typu 2 i cukrzyc monogenowych
(tab. 2.). W rejonach geograficznych, w których zapadalność na cukrzycę typu 1 jest
mniejsza, częściej pierwszym objawem jest cukrzycowa
kwasica ketonowa.38
| Tabela 3. Cechy kliniczne cukrzycy typu 1 i 2 oraz cukrzyc monogenowych u dzieci i młodzieży | |||
|---|---|---|---|
| Cechy | Cukrzyca typu 1 | Cukrzyca typu 2 | Cukrzyce monogenowe |
| dziedziczenie | wielogenowe | wielogenowe | monogenowe |
| wiek w momencie zachorowania | od 6. mż. do okresu wczesnej dorosłości | zwykle okres dojrzewania płciowego (lub później) | często po zakończeniu dojrzewania płciowego,z wyjątkiem GCK i NDM |
| objawy kliniczne | najczęściej ostre, gwałtowne | zróżnicowane, od łagodnych, o powolnym rozwoju (często podstępnym) do ciężkich | zróżnicowane(w przypadku GCK mogą być stwierdzone przypadkowo) |
| skojarzenie z: | |||
| autoimmunizacją | tak | nie | nie |
| ketozą | często | rzadko | często w NDM, rzadko w innych postaciach |
| otyłością | nie | tak | nie |
| rogowaceniem ciemnym (acanthosis nigricans) | nie | tak | nie |
| częstość (% wszystkich przypadków cukrzycy u dzieci i młodzieży) | zwykle >90% | w większości krajów<10% (w Japonii60–80%) | 1–4% |
| cukrzyca u rodzica | 2–4% | 80% | 90% |
| GCK – gen glukokinazy, NDM – cukrzyca monogenowa występującą w pierwszych6 miesiącach życia | |||
Epidemiologia cukrzycy typu 1
W większości krajów Zachodnich cukrzyca typu 1 stanowi ponad 90% wszystkich przypadków cukrzycy u dzieci i młodzieży, jednak biorąc pod
uwagę wszystkie grupy wiekowe, cukrzycę typu 1 stwierdza się u 5–10% wszystkich chorych na cukrzycę.
Ogółem szacuje się, że na całym świecie
cukrzyca typu 1 rozwija się rocznie u około 80 000
dzieci do 15. roku życia.39 Cukrzyca typu 2 występuje
coraz częściej i stanowi znaczący odsetek
zachorowań na cukrzycę u młodzieży w niektórych
populacjach dużego ryzyka,3 jednak epidemiologiczne
dane populacyjne są bardziej ograniczone
niż dane dostępne dla cukrzycy typu 1.
W starszych badaniach epidemiologicznych dotyczących
oceny zapadalności „początek cukrzycy
typu 1” jest definiowany jako data pierwszego
wstrzyknięcia insuliny, ponieważ czas od wystąpienia
pierwszych objawów do ustalenia rozpoznania
był różny,40 jednak w aktualnych wytycznych
cukrzycę definiuje się na podstawie nieprawidłowych
wyników badań laboratoryjnych (tab. 1.).
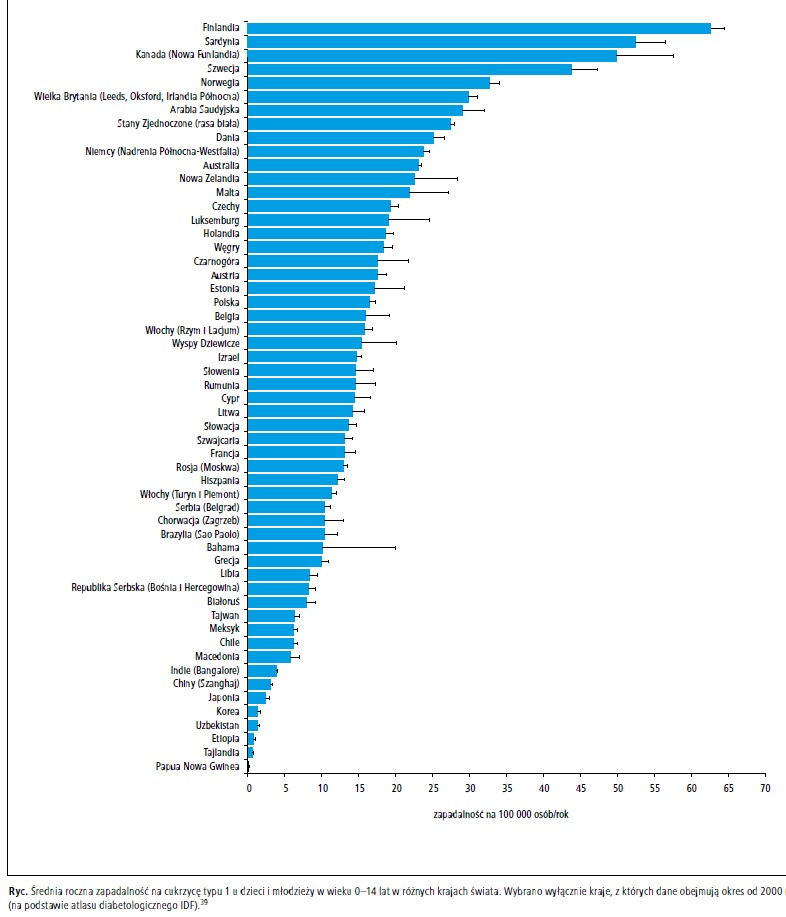
Obserwuje się duże różnice w zapadalności
na cukrzycę typu 1 pomiędzy poszczególnymi krajami, w obrębie danego kraju oraz pomiędzy od
miennymi etnicznie populacjami (ryc.), przy czym
największą zapadalność obserwuje się w Finlandii,
41 krajach Europy Północnej42-44 i Kanadzie.45 W obrębie populacji rasy białej zamieszkującej
Europę różnica w zapadalności jest blisko 20-krotna,
25 a wskaźniki zapadalności korelują z częstością
występowania w ogólnej populacji genotypu
HLA predysponującego do zachorowania.46,47
Wśród około 500 000 dzieci chorych na cukrzycę
typu 1, około 26% pochodzi z Europy, a 22% z Ameryki Północnej i regionu Karaibów.39 W krajach azjatyckich zapadalność na cukrzycę
typu 1 jest bardzo mała, w Japonii wynosi około
2/100 000 osobolat,48 w Chinach (Szanghaj)
3,1/100 000,49 na Taiwanie około 5/100 00050 i – w porównaniu z rasą białą – wykazuje związek z innymi, rzadkimi HLA.51-54 Ponadto w Japonii
występuje odmienna postać powoli postępującej
cukrzycy typu 1, która obecnie stanowi niemal 1/3
wszystkich przypadków cukrzycy typu 1 w tym
kraju.55,56 Porównanie średniego rocznego współczynnika
zapadalności na cukrzycę typu 1 u dzieci
do 15. roku życia w różnych krajach przedstawiono
na rycinie.
Dobrze opisano sezonową zmienność występowania
nowych zachorowań, ze szczytem w miesiącach
zimowych, natomiast w innych publikacjach
wykazano większą zapadalność w ciepłych
porach roku49 lub zmieniającą się sezonowość z roku na rok.57-59 Poza tym stwierdzono, że rozwój
procesu autoimmunizacyjnego obejmującego
wysepki trzustkowe również wykazuje sezonową
zmienność, podobnie jak związek pomiędzy miesiącem
urodzenia a ryzykiem cukrzycy typu 1.60-61
W przeciwieństwie do większości chorób autoimmunizacyjnych,
które zdecydowanie częściej
występują u płci żeńskiej, różnice w zapadalności
na cukrzycę typu 1 u płci męskiej i żeńskiej stwierdza
się tylko w niektórych populacjach. Na ogół
jednak w wieku młodzieńczym i u młodych dorosłych
obserwuje się tendencję do większej zachorowalności
wśród mężczyzn.59,62,63
Na przestrzeni ostatnich dziesięcioleci na całym
świecie obserwuje się zwiększenie zapadalności
na cukrzycę typu 1.41,43,49,50,57-59,64-72 Według niektórych
publikacji niewspółmiernie duże zwiększenie
liczby zachorowań dotyczy dzieci w wieku
<5 lat64-73 oraz krajów rozwijających się lub krajów, w których w ostatnich dziesięcioleciach zaszły
przemiany gospodarcze.64,68 Udokumentowano,
że w niektórych krajach zapadalność utrzymuje
się od kilku lat na stałym poziomie.41,43,69,74,75
Zwiększenie zapadalności na cukrzycę typu 1 jest
związane ze zwiększaniem się w niektórych populacjach
odsetka osób z genotypem HLA obarczonym
małym ryzykiem zachorowania.76-78 Sugeruje
to, że rozwój choroby w coraz większym stopniu
zależy od czynników środowiskowych.
Cukrzyca typu 1 występuje rodzinnie tylko w około 10% przypadków,79 a po uwzględnieniu
wywiadu dotyczącego dalszych krewnych odsetek
ten przekracza już 20%.80 Mimo to model dziedziczenia
choroby pozostaje nieznany. Ryzyko wystąpienia
cukrzycy typu 1 u jednojajowego bliźniaka
chorego nie przekracza 40%,25,81 ryzyko u rodzeństwa
wynosi niemal 4% do ukończenia 20. roku życia82,83
oraz 9,6% do ukończenia 60. roku życia,49 w porównaniu z 0,5% w populacji ogólnej. Zbiorcze
ryzyko rozwoju cukrzycy do ukończenia 15. roku
życia jest większe u rodzeństwa posiadającego
identyczny genotyp HLA DR3-DQ2/DR4-DQ8
(17 vs 6% u rodzeństwa posiadającego 1 wspólny
haplotyp lub odmienne haplotypy).84 Ryzyko jest
większe u rodzeństwa osób, u których cukrzycę
rozpoznano w młodszym wieku, jeżeli cukrzycę
rozpoznano u ojca w młodym wieku, u płci męskiej i u potomstwa osób, które zostały rodzicami w starszym wieku.82,84,85
Cukrzyca typu 1 występuje 2–3 razy częściej u potomstwa mężczyzn chorych na cukrzycę
(3,6–8,5%), w porównaniu z potomstwem kobiet
chorych na cukrzycę (1,3–3,6%).85-90 Ryzyko zbiorcze
cukrzycy typu 1 wynosi około 4% u potomstwa
chorych na cukrzycę typu 1, którą rozpoznano w wieku dorosłym (15–39 lat),91 przy czym nie
zależało ono od płci chorego rodzica.
Rycina.
Cukrzyce monogenowe
Rodzinną postać łagodnej cukrzycy przebiegającej
bez ketozy, objawiającej się w wieku młodzieńczym
lub we wczesnym wieku dorosłym,92,93 początkowo
określano jako cukrzycę typu MODY (maturity-onset
diabetes of the young). Obecnie uważa się
ją za grupę zaburzeń wywołaną przez dominujące
mutacje heterozygotyczne w obrębie genów
istotnych dla rozwoju lub funkcji komórek ß.93,94
Zgodnie z klasycznym opisem, MODY objawia się
przed ukończeniem 25. roku życia, jest dziedziczona
autosomalnie
dominująco i przebiega bez
ketozy.94,95 Wiadomo jednak, że objawy cukrzycy
typu 1, 2 i cukrzyc monogenowych w dużym
stopniu się pokrywają. Biorąc pod uwagę coraz
częstsze rozpoznawanie cukrzycy typu 2 u młodych
osób, wielu pacjentów spełnia wszystkie
„klasyczne” kryteria cukrzycy monogenowej, ale
wstępnie niekiedy rozpoznaje się u nich cukrzycę
typu 2.96 Niektóre cechy kliniczne (p. tab. 3.) powinny
wzbudzić u lekarza podejrzenie cukrzycy
monogenowej.
Aktualnie uważa się, że lepiej jest zdefiniować
cukrzycę monogenową na podstawie przynależności
do podgrupy genetycznej (p. tab. 2.).
Najczęstsza postać, znana również jako
MODY3, wiąże się z mutacją genu czynnika jądrowego
hepatocytów 1α (HNF1A) będącego czynnikiem
transkrypcyjnym. Większość pozostałych
przypadków wynika z mutacji genu glukokinazy
(GCK) i HNF4A, natomiast rzadkie postacie
wiążą się z mutacjami genów innych czynników
transkrypcyjnych, na przykład HNF1B, czynnika
promotora insuliny (IPF)-1 i NeuroD1 (tab. 2.).2,94
Więcej informacji podano w wytycznych ISPAD
dotyczących cukrzyc monogenowych.18
Różne typy cukrzyc monogenowych znacznie
się różnią w zakresie stopnia hiperglikemii, zapotrzebowania
na insulinę i ryzyka wystąpienia
późniejszych powikłań.
Swoiste badania molekularne pomagają przewidzieć
przebieg kliniczny choroby i wybrać optymalną
metodę leczenia. Umożliwiają także udzielenie
innym członkom rodziny chorych na cukrzycę porady
genetycznej oraz objęcie ich badaniami, co z kolei może doprowadzić do zmiany rozpoznania
typu cukrzycy.97
Cukrzyca noworodkowa
Cukrzyca typu 1 rzadko objawia się w 1. roku
życia, zwłaszcza przed ukończeniem 6. miesiąca
życia,98,99 a jej najbardziej prawdopodobną przyczyną u najmłodszych niemowląt jest mutacja
czynnika transkrypcyjnego FOXP3. W takich
przypadkach cukrzyca jest jednym z zaburzeń
składających się na sprzężony z chromosomem X
(immunodysregulation polyendocrinopathy enteropathy
X-linked syndrome – IPEX) zespół dysregulacji
immunologicznej z poliendokrynopatią i enteropatią.
100 Cukrzycę monogenową występującą w pierwszych 6 miesiącach życia określa się jako
NDM, chociaż u niektórych pacjentów pierwsze
objawy pojawiają się w 9.–12. miesiącu życia.101-103 U wielu dzieci rozpoznanie udaje się ustalić dopiero
po okresie noworodkowym, dlatego niektórzy
sugerują wprowadzenie alternatywnego pojęcia
„cukrzyca monogenowa niemowląt”,104 jednak nadal
powszechnie używa się terminu NDM.
Ta rzadka choroba (1/400 000 urodzeń) może
być związana z wewnątrzmacicznym zahamowaniem
wzrastania, co wynika z niedoboru insuliny w okresie prenatalnym,105,106 oraz innymi objawami
klinicznym niezwiązanymi z czynnością
wewnątrzwydzielniczą trzustki.
Prawie połowa chorych na NDM wymaga leczenia
przez całe życie w celu opanowania hiperglikemii
(utrwalona cukrzyca noworodków [permanent
neonatal diabetes mellitus – PNDM]). W pozostałych przypadkach w ciągu kilku tygodni
lub miesięcy dochodzi do remisji (przemijająca cukrzyca
noworodków [transient neonatal diabetes
mellitus – TNDM]), chociaż w późniejszym okresie
życia choroba może nawrócić.
Około 2/3 przypadków TNDM wynika z imprintingu
(naznaczenia rodzicielskiego) genów na długim
ramieniu chromosomu 6 (6q24).107,108 W pozostałych
przypadkach najczęściej stwierdza się
mutacje aktywujące genów kodujących dwie podjednostki
kanału potasowego zależnego od ATP w błonie komórek ß (KCNJ11, kodującego podjednostkę
Kir6.2 lub ABCC8, kodującego podjednostkę
SUR1).109 Chociaż w okresie niemowlęcym
cukrzyca ma charakter przemijający, u 50–60%
pacjentów rozwija się utrwalona cukrzyca, zwykle
około wieku pokwitania.100
PNDM jest związana z mutacjami aktywującymi
KCNJ11 i ABCC8111,111,112 oraz mutacjami genu
insulin (INS),113-117 a także rzadziej GCK118,119
oraz czynnika transkrypcyjnego niezbędnego
do rozwoju trzustki (PDX1).120 Poza tym u niemowląt
mogą występować różne postacie cukrzycy
związane z zespołami chorobowymi. Szczegółowe
informacje o podłożu genetycznym NDM można
znaleźć w rozdziale pt.: „Rozpoznawanie i leczenie
cukrzyc monogenowych u dzieci i młodzieży”.18
Cukrzyca mitochondrialna
Cukrzyca mitochondrialna zwykle jest związana z głuchotą czuciowo-nerwową (odbiorczą) i charakteryzuje
się postępującą, nieautoimmunizacyjną
niewydolnością komórek ß.121,122 Przekazanie
zmutowanego matczynego DNA mitochondrialnego
(mtDNA) może powodować wystąpienie cukrzycy
dziedziczonej od matki. Najczęściej stwierdza
się mutację w pozycji 3243 w genie leucyny tRNA,
prowadzącą do zamiany adeniny na guaninę.123,124
Cukrzyca mitochondrialna może mieć różny
przebieg – od ostrego początku z kwasicą ketonową
lub bez niej do stopniowo nasilających się pierwszych
objawów przypominających cukrzycę typu 2.
Na ogół choroba objawia się u młodych dorosłych,
ale może wystąpić również u dzieci i młodzieży, u których rzadziej niż u dorosłych stwierdza się
głuchotę.125
Mukowiscydoza i cukrzyca
CFRD jest najczęstszą chorobą współistniejącą
związaną z mukowiscydozą. Przyczyną CFRD
jest przede wszystkim niedobór insuliny, któremu
towarzyszy niedobór glukagonu i różnego stopnia
insulinooporność (zwłaszcza w ostrej fazie choroby,
wynikająca z zakażeń i stosowania leków, takich
jak leki rozszerzające oskrzela i glikokortykosteroidy
[GKS]). Wśród pozostałych czynników
wymienia się duże zapotrzebowanie kaloryczne,
opóźnione opróżnianie żołądka, zaburzenia motoryki
jelit i zaburzenie funkcji wątroby.126
Mukowiscydoza wiąże się z postępującym upośledzeniem
tolerancji glukozy wraz z upływem
czasu. Zaburzenia magą mieć postać przemijającej
hiperglikemii poposiłkowej, IGT lub cukrzycy.
Chociaż wczesna CFRD charakteryzuje się prawidłowym
stężeniem glukozy na czczo, to wraz z wiekiem
chorego parametr ten stopniowo się pogarsza.
CFRD najczęściej występuje w wieku młodzieńczym i we wczesnym dorosłym,127 choć może się
pojawić w każdym czasie, nawet u niemowląt.
Choroba może przebiegać bezobjawowo, skrycie.
Niekiedy jest przyczyną słabego przyrostu masy ciała, 128 a jej manifestację może przyspieszyć insulinooporność
związana z zakażeniem lub stosowaniem
GKS. Częstość rozpoznawania CFRD
zależy od stosowanych badań przesiewowych.129
Za początek CFRD uznaje się datę, kiedy chory
na mukowiscydozę spełnił kryteria diagnostyczne
cukrzycy, nawet jeśli w kolejnych dniach hiperglikemia
ustąpiła.
Wystąpienie CFRD charakteryzuje się złym
rokowaniem, ponieważ zwiększa ryzyko powikłań i umieralność, co obserwowano przed wprowadzeniem
rutynowych badań przesiewowych w kierunku CFRD i wczesnej terapii insuliną.130
Źle kontrolowana CFRD upośledza odpowiedź immunologiczną
na zakażenia i nasila katabolizm
białek.129,131
Coroczne badania przesiewowe w kierunku
CFRD należy rozpocząć do ukończenia 10. roku
życia u wszystkich chorych na mukowiscydozę, u których nie stwierdzono CFRD. W ramach badań
przesiewowych należy stosować 2-godzinny
OGTT z 75 g glukozy (1,75 g/kg mc.). Więcej informacji
można znaleźć w wytycznych ISPAD dotyczących
CFRD.132
Cukrzyca wywołana przez leki i toksyny
Wiele leków upośledza wydzielanie insuliny
(np. propranolol) i/lub jej funkcję (np. GKS, leki
przeciwpsychotyczne), a niektóre z nich (np. pentamidyna)
mogą nawet trwale uszkodzić komórki
ß.2,133,134
W neurochirurgii, w celu zapobiegania obrzękowi
mózgu, często stosuje się duże dawki deksametazonu.
Dodatkowy stres związany z operacją
może potęgować indukowaną lekiem insulinooporność,
wywołując względny niedobór insuliny, wystarczający
do wystąpienia przemijającej postaci
cukrzycy. Może się ona nasilać w przypadku leczenia
moczówki prostej dożylnymi wlewami roztworów
glukozy w dużej objętości. Dożylny wlew
insuliny jest najlepszym sposobem kontrolowania
hiperglikemii, która zwykle jest przemijająca.
W leczeniu onkologicznym stosuje się diabetogenne
leki: L-asparaginazę, GKS w dużych
dawkach, cyklosporynę lub takrolimus (FK506).
L-asparaginaza zwykle wywołuje odwracalną postać cukrzycy.135v
Takrolimus i cyklosporyna
mogą powodować wystąpienie cukrzycy utrwalonej,
prawdopodobnie na drodze niszczenia komórek
wysp trzustkowych.136 Częstym zjawiskiem
jest cykliczne występowanie cukrzycy, w czasie
kolejnych cyklów chemioterapii, zwłaszcza w przypadku
podawania GKS w dużych dawkach.
U pacjentów po przeszczepieniu narządów cukrzyca
najczęściej występuje w wyniku zastosowania
takrolimusu oraz GKS w dużych dawkach.
Ryzyko jej wystąpienia jest większe u otyłych
chorych.137-139
Cukrzycę mogą także wywoływać nietypowe
leki przeciwpsychotyczne, takie jak olanzapina,
rysperydon, kwetiapina oraz zyprazydon, co może
być związane ze zwiększeniem masy ciała. Stosowanie
leków przeciwpsychotycznych u dzieci i młodzieży
wiązało się z ponad 3-krotnie większym
ryzykiem wystąpienia cukrzycy nieautoimmunizacyjnej i było ono coraz większe po zwiększeniu
się dawki kumulacyjnej leku.140 U dzieci kanadyjskich
chorych na cukrzycę polekową rzadziej
obserwowano czynniki ryzyka cukrzycy typu 2 (dodatni wywiad rodzinny w kierunku cukrzycy,
rasa inna niż biała, rogowacenie ciemne) niż u dzieci chorych na cukrzycę typu 2.141
Hiperglikemia stresowa
Hiperglikemię stresową obserwowano u ≤5% dzieci
przyjmowanych na szpitalne oddziały ratunkowe z powodu ostrego zachorowania/posocznicy, urazów,
drgawek gorączkowych, oparzeń i podwyższonej
temperatury ciała (>39oC).142-145 Jednak
ciężką hiperglikemię (≥16,7 mmol/l lub 300 mg/dl)
stwierdzano u <1%, a prawie u 2/3 pacjentów przed
oceną glikemii zastosowano interwencję wpływającą
na stężenie glukozy, co sugeruje, że etiologia
tej postaci hiperglikemii może być przynajmniej
częściowo
jatrogenna.146
Progresja do klinicznie jawnej cukrzycy następuje w 0–32% przypadków.145,147-152 Ryzyko
rozwoju cukrzycy było większe u dzieci z przygodną
hiperglikemią bez towarzyszącej ciężkiej
choroby niż w przypadku hiperglikemii u dzieci,
które były ciężko chore.150 Jak można oczekiwać,
oznaczanie autoprzeciwciał związanych z cukrzycą
charakteryzuje się dużą wartością predykcyjną wyniku dodatniego i ujemnego w diagnostyce
cukrzycy typu 1 u dzieci z hiperglikemią stresową.
150 U dzieci, które uległy ciężkim oparzeniom,
insulinooporność może się utrzymywać do 3 lat
od zdarzenia.144
Konflikt interesów: Autorzy nie zgłosili konfliktu intetresów.
Piśmiennictwo:
1. World Health Organisation: Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycaemia: Report of a WHO/IDF Consultation. Geneva, Switzerland: World Health Organisation, 20062. American Diabetes Association: Diagnosis and classification of diabetes mellitus. Diabetes Care, 2014; 37 (suppl. 1): S81–S90
3. Zeitler P., Fu J., Tandon N., et al.: Type 2 diabetes in the child and adolescent. Pediatr. Diabetes, 2014; 15 (suppl. 20): 26–46
4. American Diabetes Association: Standards of medical care in diabetes – 2014. Diabetes Care, 2014; 37 (suppl. 1): S14–S80
5. World Health Organization: Use of Glycated Haemoglobin (HbA1c) in the Diagnosis of Diabetes Mellitus. Geneva, Switzerland: World Health Organization, 2011
6. Islam S.T., Abraham A., Donaghue K.C., et al.: Plateau of adiposity in Australian children diagnosed with type 1 diabetes: a 20-year study. Diabet. Med., 2014; 31: 686–690
7. Kapellen T.M., Gausche R., Dost A., et al.: Children and adolescents with type 1 diabetes in Germany are more overweight than healthy controls: results comparing DPV database and CrescNet database. J. Pediatr. Endocrinol. Metab., 2014; 27: 209–214
8. Rewers A., Klingensmith G., Davis C., et al.: Presence of diabetic ketoacidosis at diagnosis of diabetes mellitus in youth: the Search for Diabetes in Youth Study. Pediatrics, 2008; 121: e1258–e1266
9. Dabelea D., Rewers A., Stafford J.M., et al.: Trends in the prevalence of ketoacidosis at diabetes diagnosis: the SEARCH for diabetes in youth study. Pediatrics, 2014; 133: e938–e945
10. Fendler W., Borowiec M., Baranowska-Jazwiecka A., et al.: Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia, 2012; 55: 2631–2635
11. Irgens H.U., Molnes J., Johansson B.B., et al.: Prevalence of monogenic diabetes in the population based Norwegian Childhood Diabetes Registry. Diabetologia, 2013; 56: 1512–1519
12. Pihoker C., Gilliam L.K., Ellard S., et al.: Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for Diabetes in Youth. J. Clin. Endocrinol. Metab., 2013; 98: 4055–4062
13. Galler A., Stange T., Muller G., et al.: Incidence of childhood diabetes in children aged less than 15 years and its clinical and metabolic characteristics at the time of diagnosis: data from the Childhood Diabetes Registry of Saxony, Germany. Horm. Res. Paediatr., 2010; 74: 285–291
14. Gill G.V., Mbanya J.C., Ramaiya K.L., Tesfaye S.: A sub-Saharan African perspective of diabetes. Diabetologia, 2009; 52: 8–16
15. Barman K.K., Premalatha G., Mohan V.: Tropical chronic pancreatitis. Postgrad. Med. J., 2003; 79: 606–615
16. Watkins R.A., Evans-Molina C., Blum J.S., Dimeglio L.A.: Established and emerging biomarkers for the prediction of type 1 diabetes: a systematic review. Transl. Res., 2014; 164: 110–121
17. Dabelea D., Pihoker C., Talton J.W., et al.: Etiological approach to characterization of diabetes type: the SEARCH for Diabetes in Youth Study. Diabetes Care, 2011; 34: 1628–1633
18. Rubio-Cabezas O., Hattersley A., Njolstad P., et al.: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr. Diabetes, 2014; 15 (Suppl. 20): 47–64
19. Howson J.M., Stevens H., Smyth D.J., et al.: Evidence that HLA class I and II associations with type 1 diabetes, autoantibodies to GAD and autoantibodies to IA-2, are distinct. Diabetes, 2011; 60: 2635–2644
20. Barrett J.C., Clayton D.G., Concannon P., et al.: Genome-wide association study andmeta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet., 2009; 41: 703–707
21. Noble J.A., Valdes A.M., Cook M., Klitz W., Thomson G., Erlich H.A.: The role of HLA class II genes in insulin-dependent diabetesmellitus: molecular analysis of 180 Caucasian, multiplex families. Am. J. Hum. Genet., 1996; 59: 1134–1148
22. Lambert A.P., Gillespie K.M., Thomson G., et al.: Absolute risk of childhood-onset type 1 diabetes defined by human leukocyte antigen class II genotype: a population-based study in the United Kingdom. J. Clin. Endocrinol. Metab., 2004; 89: 4037–4043
23. Nguyen C., Varney M.D., Harrison L.C., Morahan G.: Definition of high-risk type 1 diabetes HLA-DR and HLA-DQ types using only three single nucleotide polymorphisms. Diabetes, 2013; 62: 2135–2140
24. Erlich H., Valdes A.M., Noble J., et al.: HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes, 2008; 57: 1084–1092
25. Knip M.: Pathogenesis of type 1 diabetes: implications for incidence trends. Horm. Res. Paediatr., 2011; 76 (suppl. 1): 57–64
26. Aly T.A., Ide A., Jahromi M.M., et al.: Extreme genetic risk for type 1A diabetes. Proc. Natl. Acad. Sci. U S A, 2006; 103: 14 074–14 079
27. Steck A.K., Wong R., Wagner B., et al.: Effects of non-HLA gene polymorphisms on development of islet autoimmunity and type 1 diabetes in a population with high-risk HLA-DR, DQ genotypes. Diabetes, 2012; 61: 753–758
28. Ziegler A.G., Rewers M., Simell O., et al.: Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA, 2013; 309: 2473–2479
29. Bonifacio E., Krumsiek J., Winkler C., Theis F.J., Ziegler A.G.: A strategy to find gene combinations that identify children who progress rapidly to type 1 diabetes after islet autoantibody seroconversion. Acta Diabetol., 2014; 51: 403–411
30. DPT-1 Study Group: Effects of insulin in relatives of patients with type 1 diabetes mellitus. N. Engl. J. Med., 2002; 346: 1685–1691
31. Verge C.F., Gianani R., Kawasaki E., et al.: Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes, 1996; 45: 926–933
32. Skyler J.S., Krischer J.P., Wolfsdorf J., et al.: Effects of oral insulin in relatives of patients with type 1 diabetes: the diabetes prevention trial – type 1. Diabetes Care, 2005; 28: 1068–1076
33. Yeung G., Rawlinson W.D., Craig M.E.: Enterovirus infection and type 1 diabetes mellitus – a systematic review of molecular studies. BMJ, 2011; 342: d35
34. Laitinen O.H., Honkanen H., Pakkanen O., et al.: Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes, 2014; 63: 446–455
35. Richardson S.J., Willcox A., Bone A.J., Foulis A.K., Morgan N.G.: The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia, 2009; 52: 1143–1151
36. Dotta F., Censini S., van Halteren A.G., et al.: Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc. Natl. Acad. Sci. U S A, 2007; 104: 5115–5120
37. Richardson S.J., Leete P., Bone A.J., Foulis A.K., Morgan N.G.: Expression of the enteroviral Capsie protein VP1 in the islet cells of patients with type 1 diabetes is associated with induction of protein kinase R and down regulation of Mcl-1. Diabetologia, 2013; 56: 185–193
38. Usher-Smith J.A., Thompson M.J., Sharp S.J., Walter F.M.: Factors associated with the presence of diabetic ketoacidosis at diagnosis of diabetes in children and young adults: a systematic review. BMJ, 2011; 343: d4092
39. International Diabetes F. IDF Diabetes Atlas. 6th edn. Brussels, Belgium: International Diabetes F, 2013
40. Diamond Project Group: Incidence and trends of childhood type 1 diabetes worldwide 1990–1999. Diabet. Med., 2006; 23: 857–866
41. Harjutsalo V., Sund R., Knip M., Groop P.H.: Incidence of type 1 diabetes in Finland. JAMA, 2013; 310: 427–428
42. Berhan Y., Waernbaum I., Lind T., Mollsten A., Dahlquist G., Swedish Childhood Diabetes Study Group: Thirty years of prospective nationwide incidence of childhood type 1 diabetes: the accelerating increase by time tends to level off in Sweden. Diabetes, 2011; 60: 577–581
43. Skrivarhaug T., Stene L.C., Drivvoll A.K., Strom H., Joner G., Norwegian Childhood Diabetes Study Group: Incidence of type 1 diabetes in Norway among children aged 0–14 years between 1989 and 2012 has the incidence stopped rising? Results from the Norwegian Childhood Diabetes Registry. Diabetologia, 2014; 57: 57–62
44. Rawshani A., Landin-Olsson M., Svensson A.M., et al.: The incidence of diabetes among 0–34 year olds in Sweden: new data and better methods. Diabetologia, 2014; 57: 1375–1381
45. Newhook L.A., Penney S., Fiander J., Dowden J.: Recent incidence of type 1 diabetes mellitus in children 0–14 years in Newfoundland and Labrador, Canada climbs to over 45/100,000: a retrospective time trend study. BMC Res. Notes, 2012; 5: 628
46. Ilonen J., Reijonen H., Green A., et al.: Geographical differences within finland in the frequency of HLADQ genotypes associated with type 1 diabetes susceptibility. Eur. J. Immunogenet., 2000; 27: 225–230
47. Kukko M., Virtanen S.M., Toivonen A., et al.: Geographical variation in risk HLA-DQB1 genotypes for type 1 diabetes and signs of beta-cell autoimmunity in a high-incidence country. Diabetes Care, 2004; 27: 676–681
48. Tajima N., Morimoto A.: Epidemiology of childhood diabetes mellitus in Japan. Pediatr. Endocrinol. Rev., 2012; 10 (suppl. 1): 44–50
49. Zhao Z., Sun C., Wang C., et al.: Rapidly rising incidence of childhood type 1 diabetes in Chinese population: epidemiology in Shanghai during 1997–2011. Acta Diabetol., 2014 9Epub ahead of print)
50. Lin W.H., Wang M.C., Wang W.M., et al.: Incidence of and mortality from type I diabetes in Taiwan from 1999 through 2010: a nationwide cohort study. PLoS One, 2014; 9: e86 172
51. Park Y.: Why is type 1 diabetes uncommon in Asia? Ann. N Y Acad. Sci., 2006; 1079: 31–40
52. Park Y.S., Wang C.Y., Ko K.W., et al.: Combinations of HLA DR and DQ molecules determine the susceptibility to insulin-dependent diabetes mellitus in Koreans. Hum. Immunol., 1998; 59: 794–801
53. Ikegami H., Fujisawa T., Kawabata Y., Noso S., Ogihara T.: Genetics of type 1 diabetes: similarities and differences between Asian and Caucasian populations. Ann. N Y Acad. Sci., 2006; 1079: 51–59
54. Sugihara S.: Genetic susceptibility of childhood type 1 diabetes mellitus in Japan. Pediatr. Endocrinol. Rev., 2012; 10 (suppl. 1): 62–71
55. Urakami T., Suzuki J., Yoshida A., Saito H., Mugishima H.: Incidence of children with slowly progressive form of type 1 diabetes detected by the urine glucose screening at schools in the Tokyo metropolitan area. Diabetes Res. Clin. Pract., 2008; 80: 473–476
56. Urakami T., Yoshida A., Suzuki J., et al.: Differences in prevalence of antibodies to GAD and IA-2 and their titers at diagnosis in children with slowly and rapidly progressive forms of type 1 diabetes. Diabetes Res. Clin. Pract., 2009; 83: 89–93
57. Imkampe A.K., Gulliford M.C.: Trends in type 1 diabetes incidence in the UK in 0- to 14-year-olds and in 15- to 34-year-olds, 1991–2008. Diabet. Med.,. 2011; 28: 811–814
58. Jarosz-Chobot P., Polańska J., Szadkowska A., et al.: Rapid increase in the incidence of type 1 diabetes in Polish children from 1989 to 2004, and predictions for 2010 to 2025. Diabetologia, 2011; 54: 508–515
59. Skordis N., Efstathiou E., Kyriakides T.C., et al.: Epidemiology of type 1 diabetes mellitus in Cyprus: rising incidence at the dawn of the 21st century. Hormones (Athens), 2012; 11: 86–93
60. Laron Z., Lewy H., Wilderman I., et al.: Seasonality of month of birth of children and adolescents with type 1 diabetes mellitus in homogenous and heterogeneous populations. Isr. Med. Assoc. J., 2005; 7: 381–384
61. Kahn H.S., Morgan T.M., Case L.D., et al.: Association of type 1 diabetes with month of birth among U.S. youth: the SEARCH for Diabetes in Youth Study. Diabetes Care, 2009; 32: 2010–2015
62. Weets I., Kaufman L., Van der Auwera B., et al.: Seasonality in clinical onset of type 1 diabetes in Belgian patients above the age of 10 is restricted to HLA-DQ2/DQ8-negative males, which explains the male to female excess in incidence. Diabetologia, 2004; 47: 614–621
63. Wandell P.E., Carlsson A.C.: Time trends and gender differences in incidence and prevalence of type 1 diabetes in Sweden. Curr. Diabetes Rev., 2013; 9: 342–349
64. Patterson C.C., Dahlquist G.G., Gyurus E., Green A., Soltesz G.: Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: a multicentre prospective registration study. Lancet, 2009; 373: 2027–2033
65. Schober E., Waldhoer T., Rami B., Hofer S.: Incidence and time trend of type 1 and type 2 diabetes in Austrian children 1999–2007. J. Pediatr., 2009; 155: 190–193 e1
66. Haynes A., Bulsara M.K., Bower C., Jones T.W., Davis E.A.: Cyclical variation in the incidence of childhood type 1 diabetes in Western Australia (1985–2010). Diabetes Care, 2012; 35: 2300–2302
67. Derraik J.G., Reed P.W., Jefferies C., Cutfield S.W., Hofman P.L., Cutfield W.S.: Increasing incidence and age at diagnosis among children with type 1 diabetes mellitus over a 20-year period in Auckland (New Zealand). PLoS One, 2012; 7: e32 640
68. Sipetic S., Maksimovic J., Vlajinac H., et al.: Rising incidence of type 1 diabetes in Belgrade children aged 0–14 years in the period from 1982 to 2005. J. Endocrinol. Invest., 2013; 36: 307–312
69. Bruno G., Maule M., Biggeri A., et al.: More than 20 years of registration of type 1 diabetes in Sardinian children: temporal variations of incidence with age, period of diagnosis, and year of birth. Diabetes, 2013; 62: 3542–3546
70. Lipman T.H., Levitt Katz L.E., Ratcliffe S.J., et al.: Increasing incidence of type 1 diabetes in youth: twenty years of the Philadelphia Pediatric Diabetes Registry. Diabetes Care, 2013; 36: 1597–1603
71. Tran F., Stone M., Huang C.Y., et al.: Populationbased incidence of diabetes in Australian youth aged 10–18 yr: increase in type 1 diabetes but not type 2 diabetes. Pediatr. Diabetes, 2014 Mar 17. doi: 10.1111/pedi.12 131. (Epub ahead of print)
72. Lawrence J.M., Imperatore G., Dabelea D., et al.: Trends in incidence of type 1 diabetes among non-Hispanic White youth in the United States, 2002–2009. Diabetes, 2014 Jun 4. pii: DB_131 891. (Epub ahead of print)
73. Gyurus E.K., Patterson C., Soltesz G.: Twenty-one years of prospective incidence of childhood type 1 diabetes in Hungary – the rising trend continues (or peaks and highlands?). Pediatr. Diabetes, 2012; 13: 21–25
74. Cinek O., Kulich M., Sumnik Z.: The incidence of type 1 diabetes in young Czech children stopped rising. Pediatr. Diabetes, 2012; 13: 559–563
75. Kraan J.A., Claessen F.M.A.P., Elliott K.D., et al.: Population based incidence of type 1 diabetes in New South Wales Australia 1990–2010 – have we reached a plateau? (Oral Abstract). Pediatr. Diabetes, 2011; 12 (suppl. 15): 25
76. Hermann R., Knip M., Veijola R., et al.: Temporal changes in the frequencies of HLA genotypes in patients with type 1 diabetes – indication of an increased environmental pressure? Diabetologia, 2003; 46: 420–425
77. Gillespie K.M., Bain S.C., Barnett A.H., et al.: The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes. Lancet, 2004; 364: 1699–1700
78. Fourlanos S., Varney M.D., Tait B.D., et al.: The rising incidence of type 1 diabetes is accounted for by cases with lower-risk human leukocyte antigen genotypes. Diabetes Care, 2008; 31: 1546–1549
79. Hemminki K., Li X., Sundquist J., Sundquist K.: Familial association between type 1 diabetes and other autoimmune and related diseases. Diabetologia, 2009; 52: 1820–1828
80. Parkkola A., Harkonen T., Ryhanen S.J., Ilonen J., Knip M.: Extended family history of type 1 diabetes and phenotype and genotype of newly diagnosed children. Diabetes Care, 2012; 36: 348–354
81. Olmos P., A’Hern R., Heaton D.A., et al.: The significance of the concordance rate for type 1 (insulindependent) diabetes in identical twins. Diabetologia, 1988; 31: 747–750
82. Harjutsalo V., Podar T., Tuomilehto J.: Cumulative incidence of type 1 diabetes in 10,168 siblings of Finnish young-onset type 1 diabetic patients. Diabetes, 2005; 54: 563–569
83. Steck A.K., Barriga K.J., Emery L.M., Fiallo-Scharer R.V., Gottlieb P.A., Rewers M.J.: Secondary attack rate of type 1 diabetes in Colorado families. Diabetes Care, 2005; 28: 296–300
84. Gillespie K.M., Aitken R.J., Wilson I., Williams A.J., Bingley P.J.: Early onset of diabetes in the proband is themajor determinant of risk inHLADR3-DQ2/DR4-DQ8 siblings. Diabetes, 2014; 63: 1041–1047
85. Gillespie K.M., Gale E.A., Bingley P.J.: High familial risk and genetic susceptibility in early onset childhood diabetes. Diabetes, 2002; 51: 210–214
86. Green A., Schober E., Christov V., et al.: Familial risk of type 1 diabetes in European children. Diabetologia, 1998; 41: 1151–1156
87. Dorman J.S., Steenkiste A.R., O’Leary L.A., McCarthy B.J., Lorenzen T., Foley T.P.: Type 1 diabetes in offspring of parents with type 1 diabetes: the tip of an autoimmune iceberg? Pediatr. Diabetes, 2000; 1: 17–22
88. El Hashimy M., Angelico M.C., Martin B.C., Krolewski A.S., Warram J.H.: Factors modifying the risk of IDDM in offspring of an IDDM parent. Diabetes, 1995; 44: 295–299
89. Lorenzen T., Pociot F., Stilgren L., et al.: Predictors of IDDM recurrence risk in offspring of Danish IDDM patients. Danish IDDM Epidemiology and Genetics Group. Diabetologia, 1998; 41: 666–673
90. Warram J.H., Krolewski A.S., Gottlieb M.S., Kahn C.R.: Differences in risk of insulin-dependent diabetes in offspring of diabetic mothers and diabetic fathers. N. Engl. J. Med., 1984; 311: 149–152
91. Harjutsalo V., Lammi N., Karvonen M., Groop P.H.: Age at onset of type 1 diabetes in parents and recurrence risk in offspring. Diabetes, 2010; 59: 210–214
92. Tattersall R.: Maturity-onset diabetes of the young: a clinical history. Diabet. Med., 1998; 15: 11–14
93. Fajans S.S., Bell G.I.: MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care, 2011; 34: 1878–1884
94. Fajans S.S., Bell G.I., Polonsky K.S.: Molecular mechanisms and clinical pathophysiology of maturityonset diabetes of the young. N. Engl. J. Med., 2001; 345: 971–980
95. Tattersall R.B., Fajans S.S.: A difference between the inheritance of classical juvenile-onset and maturityonset type diabetes of young people. Diabetes, 1975; 24: 44–53
96. Awa W.L., Schober E., Wiegand S., et al.: Reclassification of diabetes type in pediatric patients initially classified as type 2 diabetes mellitus: 15 years followup using routine data from the German/Austrian DPV database. Diabetes Res. Clin. Pract., 2011; 94: 463–467
97. Murphy R., Ellard S., Hattersley A.T.: Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat. Clin. Pract. Endocrinol. Metab., 2008; 4: 200–213
98. Edghill E.L., Dix R.J., Flanagan S.E., et al.: HLA genotyping supports a nonautoimmune etiology in patients diagnosed with diabetes under the age of 6 months. Diabetes, 2006; 55: 1895–1898
99. Iafusco D., Stazi M.A., Cotichini R., et al.: Permanent diabetes mellitus in the first year of life. Diabetologia, 2002; 45: 798–804
100. Rubio-Cabezas O., Minton J.A., Caswell R., et al.: Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care, 2009; 32: 111–116
101. Rubio-Cabezas O., Flanagan S.E., Damhuis A., Hattersley A.T., Ellard S.: KATP channel mutations in infants with permanent diabetes diagnosed after 6 months of life. Pediatr. Diabetes, 2012; 13: 322–325
102. Rubio-Cabezas O., Edghill E.L., Argente J., Hattersley A.T.: Testing for monogenic diabetes among children and adolescents with antibodynegative clinically defined Type 1 diabetes. Diabet. Med., 2009; 26: 1070–1074
103. Mohamadi A., Clark L.M., Lipkin P.H., Mahone E.M., Wodka E.L., Plotnick L.P.: Medical and developmental impact of transition from subcutaneous insulin to oral glyburide in a 15-yr-old boy with neonatal diabetes mellitus and intermediateDENDsyndrome: extending the age of KCNJ11 mutation testing in neonatal DM. Pediatr. Diabetes, 2010; 11: 203–207
104. Massa O., Iafusco D., D’Amato E., et al.: KCNJ11 activating mutations in Italian patientswith permanent neonatal diabetes. Hum. Mutat., 2005; 25: 22–27
105. Iafusco D., Massa O., Pasquino B., et al.: Minimal incidence of neonatal/infancy onset diabetes in Italy is 1:90,000 live births. Acta Diabetol., 2012; 49: 405–408
106. von Muhlendahl K.E., Herkenhoff H.: Long-term course of neonatal diabetes. N. Engl. J. Med., 1995; 333: 704–708
107. Temple I.K., Gardner R.J., Mackay D.J., Barber J.C., Robinson D.O., Shield J.P.: Transient neonatal diabetes: widening the understanding of the etiopathogenesis of diabetes. Diabetes, 2000; 49: 1359–1366
108. Gardner R.J., Mackay D.J., Mungall A.J., et al.: An imprinted locus associated with transient neonatal diabetes mellitus. Hum. Mol. Genet., 2000; 9: 589–596
109. Flanagan S.E., Patch A.M., Mackay D.J., et al.: Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes, 2007; 56: 1930–1937
110. Temple I.K., Shield J.P.: 6q24 transient neonatal diabetes. Rev. Endocr. Metab. Disord., 2010; 11: 199–204
111. Gloyn A.L., Cummings E.A., Edghill E.L., et al.: Permanent neonatal diabetes due to paternal germline mosaicism for an activating mutation of the KCNJ11 Gene encoding the Kir6.2 subunit of the beta-cell potassium adenosine triphosphate channel. J. Clin. Endocrinol. Metab., 2004; 89: 3932–3935
112. Massa O., Iafusco D., D’Amato E., et al.: KCNJ11 activating mutations in Italian patients with permanent neonatal diabetes. Hum. Mutat., 2005; 25: 22–27
113. Colombo C., Porzio O., Liu M., et al.: Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J. Clin. Invest., 2008; 118: 2148–2156
114. Edghill E.L., Flanagan S.E., Patch A.M., et al.: Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes, 2008; 57: 1034–1042
115. Polak M., Dechaume A., Cave H., et al.: Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy: a report from the French ND (Neonatal Diabetes) Study Group. Diabetes, 2008; 57: 1115–1119
116. Stoy J., Edghill E.L., Flanagan S.E., et al.: Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. U S A, 2007; 104: 15 040–15 044
117. Garin I., Edghill E.L., Akerman I., et al.: Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc. Natl. Acad. Sci. U S A, 2010; 107: 3105–3110
118. Njolstad P.R., Sovik O., Cuesta-Munoz A., et al.: Neonatal diabetes mellitus due to complete glucokinase deficiency. N. Engl. J. Med., 2001; 344: 1588–1592
119. Rubio-Cabezas O., Ellard S.: Diabetes mellitus in neonates and infants: genetic heterogeneity, clinical approach to diagnosis, and therapeutic options. Horm. Res. Paediatr., 2013; 80: 137–146
120. Stoffers D.A., Zinkin N.T., Stanojevic V., Clarke W.L., Habener J.F.: Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet., 1997; 15: 106–110
121. Guillausseau P.J., Dubois-Laforgue D., Massin P., et al.: Heterogeneity of diabetes phenotype in patients with 3243 bp mutation of mitochondrial DNA (maternally inherited diabetes and deafness or MIDD). Diabetes Metab., 2004; 30: 181–186
122. Laloi-Michelin M., Meas T., Ambonville C., et al.: The clinical variability of maternally inherited diabetes and deafness is associated with the degree of heteroplasmy in blood leukocytes. J. Clin. Endocrinol. Metab., 2009; 94: 3025–3030
123. Reardon W., Ross R.J., Sweeney M.G., et al.: Diabetes mellitus associated with a pathogenic point mutation in mitochondrial DNA. Lancet, 1992; 340: 1376–1379
124. van den Ouweland J.M., Lemkes H.H., Ruitenbeek W., et al.: Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet., 1992; 1: 368–371
125. Mazzaccara C., Iafusco D., Liguori R., et al.: Mitochondrial diabetes in children: seek and you will find it. PLoS One, 2012; 7: e34 956
126. Rana M., Munns C.F., Selvadurai H., Donaghue K.C., Craig M.E.: Cystic fibrosis-related diabetes in childrengaps in the evidence? Nat. Rev. Endocrinol., 2010; 6: 371–378
127. Rana M., Munns C.F., Selvadurai H.C., et al.: Increased detection of cystic-fibrosis-related diabetes in Australia. Arch. Dis. Child., 2011; 96: 823–826
128. Hameed S., Morton J.R., Jaffe A., et al.: Early glucose abnormalities in cystic fibrosis are preceded by poor weight gain. Diabetes Care, 2010; 33: 221–226
129. Waugh N., Royle P., Craigie I., et al.: Screening for cystic fibrosis-related diabetes: a systematic review. Health Technol. Assess, 2012; 16 iii–iv, 1–179
130. Moran A., Dunitz J., Nathan B., Saeed A., Holme B., Thomas W.: Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care, 2009; 32: 1626–1631
131. Moran A., Milla C., Ducret R., Nair K.S.: Protein metabolism in clinically stable adult cystic fibrosis patients with abnormal glucose tolerance. Diabetes, 2001; 50: 1336–1343
132. Moran A., Pillay K., Becker D., Acerini C.L.: Management of cystic fibrosis related diabetes in children and adolescents. Pediatr. Diabetes, 2014; 15 (suppl. 20): 65–76
133. Berne C., Pollare T., Lithell H.: Effects of antihypertensive treatment on insulin sensitivity with special reference to ACE inhibitors. Diabetes Care, 1991; 14 (suppl. 4): 39–47
134. Vestri H.S., Maianu L., Moellering D.R., Garvey W.T.: Atypical antipsychotic drugs directly impair insulin action in adipocytes: effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacology, 2007; 32: 765–772
135. Pui C.H., Burghen G.A., Bowman W.P., Aur R.J.: Risk factors for hyperglycemia in children with leukemia receiving L-asparaginase and prednisone. J. Pediatr., 1981; 99: 46–50
136. Drachenberg C.B., Klassen D.K., Weir M.R., et al.: Islet cell damage associated with tacrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation, 1999; 68: 396–402
137. Al Uzri A., Stablein D.M., Cohn A.: Posttransplant diabetes mellitus in pediatric renal transplant recipients: a report of the North American Pediatric Renal Transplant Cooperative Study (NAPRTCS). Transplantation, 2001; 72: 1020–1024
138. Maes B.D., Kuypers D., Messiaen T., et al.: Posttransplantation diabetes mellitus in FK-506-treated renal transplant recipients: analysis of incidence and risk factors. Transplantation, 2001; 72: 1655–1661
139. First M.R., Gerber D.A., Hariharan S., Kaufman D.B., Shapiro R.: Posttransplant diabetes mellitus in kidney allograft recipients: incidence, risk factors, and management. Transplantation, 2002; 73: 379–386
140. Bobo W.V., Cooper W.O., Stein C.M., et al.: Antipsychotics and the risk of type 2 diabetes mellitus in children and youth. JAMA Psychiatry, 2013; 70: 1067–1075
141. Amed S., Dean H., Sellers E.A., et al.: Risk factors for medication-induced diabetes and type 2 diabetes. J. Pediatr., 2011; 159: 291–296
142. Bhisitkul D.M., Morrow A.L., Vinik A.I., Shults J., Layland J.C., Rohn R.: Prevalence of stress hyperglycemia among patients attending a pediatric emergency department. J. Pediatr., 1994; 124: 547–551
143. Valerio G., Franzese A., Carlin E., Pecile P., Perini R., Tenore A.: High prevalence of stress hyperglycaemia in children with febrile seizures and traumatic injuries. Acta Paediatr., 2001; 90: 618–622
144. Gauglitz G.G., Herndon D.N., Kulp G.A., Meyer W.J. III, Jeschke M.G.: Abnormal insulin sensitivity persists up to three years in pediatric patients post-burn. J. Clin. Endocrinol. Metab., 2009; 94: 1656–1664
145. Saz E.U., Ozen S., Simsek Goksen D., Darcan S.: Stress hyperglycemia in febrile children: relationship to prediabetes. Minerva Endocrinol., 2011; 36: 99–105
146. Weiss S.L., Alexander J., Agus M.S.: Extreme stress hyperglycemia during acute illness in a pediatric emergency department. Pediatr. Emerg. Care, 2010; 26: 626–632
147. Herskowitz R.D., Wolfsdorf J.I., Ricker A.T., et al.: Transient hyperglycemia in childhood: identification of a subgroup with imminent diabetes mellitus. Diabetes Res., 1988; 9: 161–167
148. Schatz D.A., Kowa H., Winter W.E., Riley W.J.: Natural history of incidental hyperglycemia and glycosuria of childhood. J. Pediatr., 1989; 115: 676–680
149. Vardi P., Shehade N., Etzioni A., et al.: Stress hyperglycemia in childhood: a very high risk group for the development of type I diabetes. J. Pediatr., 1990; 117: 75–77
150. Herskowitz-Dumont R., Wolfsdorf J.I., Jackson R.A., Eisenbarth G.S.: Distinction between transient hyperglycemia and early insulin-dependent diabetes mellitus in childhood: a prospective study of incidence and prognostic factors. J. Pediatr., 1993; 123: 347–354
151. Bhisitkul D.M., Vinik A.I., Morrow A.L., et al.: Prediabetic markers in children with stress hyperglycemia. Arch. Pediatr. Adolesc. Med., 1996; 150: 936–941
152. Shehadeh N., On A., Kessel I., et al.: Stress hyperglycemia and the risk for the development of type 1 diabetes. J. Pediatr. Endocrinol. Metab., 1997; 10: 283–286














