Definicja
Cukrzyca monogenowa jest wynikiem dziedziczenia jednej lub wielu mutacji w obrębie jednego genu. Może być dziedziczona dominująco, recesywnie lub być wynikiem nowo powstałej mutacji powodującej wystąpienie przypadku sporadycznego. Niemal wszystkie cukrzyce monogenowe u dzieci są wynikiem mutacji genów regulujących funkcje komórek β, chociaż rzadko cukrzyca może wystąpić na skutek mutacji powodującej bardzo ciężką insulinooporność (C).[1]
Rozpoznanie
Dlaczego należy diagnozować cukrzyce monogenowe?
U większości chorych z potwierdzoną genetycznie cukrzycą monogenową początkowo błędnie rozpoznawana jest cukrzyca typu 1 lub 2 (C).[2] Poprawne zdiagnozowanie cukrzycy monogenowej ma ważne znaczenie, ponieważ pozwala przewidzieć przebieg kliniczny choroby, wyjaśnić obecność innych związanych z nią objawów klinicznych, a co najważniejsze – wprowadzić najbardziej odpowiednie leczenie. Ponadto postawienie rozpoznania niesie konsekwencje dla innych członków rodziny, często powodując zmianę diagnozy i leczenia innych krewnych z cukrzycą oraz umożliwiając właściwe poradnictwo genetyczne.
Objawy kliniczne cukrzycy monogenowej
Do objawów klinicznych, w przypadku których powinno rozważyć się rozpoznanie cukrzycy monogenowej u dzieci, należą:
– cukrzyca noworodkowa i cukrzyca rozpoznana w pierwszych 6 miesiącach życia
– cukrzyca występująca rodzinnie, w tym u rodzica pacjenta
– łagodna (5,5–8,5 mmol/l) hiperglikemia na czczo, szczególnie stwierdzana w młodym wieku lub występująca rodzinnie
– cukrzyca związana z objawami pozatrzustkowymi.
Kiedy należy podejrzewać, że rozpoznanie cukrzycy typu 1 u dzieci może być błędne?
Objawy występujące u dzieci z początkowo rozpoznaną cukrzycą typu 1, które powinny zasugerować możliwość rozpoznania cukrzycy monogenowej, wymieniono poniżej. Żaden z nich nie ma bezwzględnej wartości diagnostycznej i powinny one być analizowane łącznie, a nie w oderwaniu od siebie (C).[3]
Kiedy należy podejrzewać, że rozpoznanie cukrzycy typu 2 u dzieci może być błędne?
Objawy występujące u dzieci, u których początkowo rozpoznano cukrzycę typu 2, a które powinny zasugerować możliwość rozpoznania cukrzycy monogenowej, wymieniono poniżej. Należy zwrócić uwagę, że większość młodocianych chorych na cukrzycę typu 2 spełnia kryteria dawnej klasyfikacji cukrzycy MODY (rozpoznanie <25 lat, dziedziczenie autosomalnie dominujące, brak insulinozależności (C).[9-12]
Postawienie rozpoznania cukrzycy monogenowej
Osoba, u której rozpoznano cukrzycę monogenową, powinna wykazywać zarówno objawy nietypowe dla cukrzycy typu 1 i 2, jak również objawy swoiste dla podtypu genetycznego cukrzycy monogenowej (E). Objawy najczęściej występujących cukrzyc monogenowych podano poniżej.
Podczas gdy w przypadku cukrzycy typu 1 i 2 nie istnieje pojedynczy test potwierdzający rozpoznanie, w >80% przypadków cukrzyc monogenowych możliwa jest diagnostyka molekularna za pomocą badania DNA (C). Diagnostyka molekularna jest dostępna w większości krajów europejskich i USA, a liczne laboratoria przeprowadzają badania chorych z innych krajów (patrz na przykład: www.diabetesgenes.org oraz www.mody.no). Badania te są kosztowne (do 500 EUR/600 USD), ale mogą mieć duży wpływ na leczenie badanej osoby i pozostałych członków rodziny, u których w przypadku zidentyfikowania mutacji badania będą tańsze (100 EUR/120 USD). Badanie niektórych spośród ostatnio opisanych mutacji genów powodujących cukrzyce monogenowe, takich jak Kir6.2 u chorych, u których rozpoznanie postawiono przed ukończeniem 6. miesiąca życia, mogą być dostępne za darmo w ramach prowadzonych badań naukowych (patrz: www.diabetesgenes.org). Tam, gdzie jest to wymagane, przed wysłaniem DNA do badania należy uzyskać zgodę ubezpieczyciela pacjenta.
W przypadku dysponowania ograniczonymi środkami ważne jest, aby badania wykonywać w sytuacjach, w których prawdopodobne jest uzyskanie wyniku pozytywnego, zmieniającego sposób leczenia. Obejmuje to dokładną selekcję kliniczną i wykonanie badań, takich jak oznaczenie stężenia peptydu C i autoprzeciwciał oraz badania innych członków rodziny przed wykonaniem badań genetycznych (E).
Szczególne podtypy cukrzyc monogenowych i ich leczenie
Cukrzyca noworodków i cukrzyca rozpoznana w pierwszych 6 miesiącach życia
Istnieją wiarygodne dowody, że cukrzyca rozpoznana w pierwszych 6 miesiącach życia nie jest cukrzycą typu 1, ponieważ u takich chorych rzadko występują autoprzeciwciała, a genotyp HLA ma w rzeczywistości charakter chroniący przed cukrzycą typu 1 (B).[41] Cukrzyca noworodków jest kolejnym zagadnieniem, które przesunęło się z klasyfikacji klinicznej do genetycznej.[13,14] Cukrzyca noworodków jest cukrzycą insulinozależną, którą zwykle rozpoznaje się w pierwszych 3 miesiącach życia. Klinicznie wyróżnia się dwie podgrupy: przemijającą cukrzycę noworodków (transient neonatal diabetes mellitus – TNDM) ustępującą w wieku średnio 12 tygodni i od tego czasu niewymagającą leczenia, chociaż aż w 50% przypadków ostatecznie dochodzi do nawrotu choroby (B).[15,16] W przeciwieństwie do tego, utrwalona cukrzyca noworodków (permanent neonatal diabetes mellitus – PNDM) wymaga ciągłego leczenia od momentu postawienia rozpoznania. U większości chorych z obydwoma rodzajami cukrzycy noworodków można określić przyczynę genetyczną. U większości chorych z TNDM występują nieprawidłowości o typie imprintingu genów ZAC i HYAMI na chromosomie 6q (B),[14,15] podczas gdy najczęstszą znaną przyczyną PNDM jest mutacja genu KCNJ11 kodującego podjednostkę Kir6.2 zależnego od ATP kanału potasowego komórek β (B).[17,18] W przypadku gdy u obojga rodziców występuje upośledzona tolerancja glukozy, najczęstszą przyczyną są homozygotyczne lub złożone heterozygotyczne mutacje glukokinazy.[19,20]
Diagnostyka różnicowa
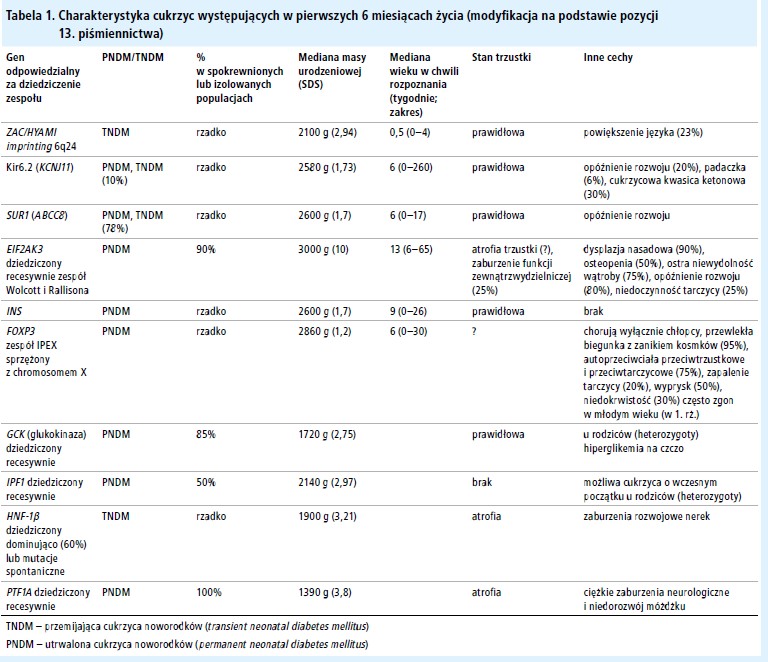
W przypadku rozpoznania cukrzycy w okresie noworodkowym trudno określić, czy jest to postać przemijająca czy utrwalona, chociaż objawy wymienione w tabeli 1 mogą pomóc w różnicowaniu podtypów i mogą być użyte do zaplanowania genetycznej diagnostyki molekularnej.
Przemijająca cukrzyca noworodków spowodowana zaburzeniem o typie imprintingu locus 6q24
Zaburzenia o typie imprintingu locus 6q24 zawierającego geny ZAC oraz HYAMI są najczęstszą przyczyną cukrzycy noworodków powodującą wystąpienie TNDM (B).[14,15,21] Do najczęstszych nieprawidłowości locus 6q24 należą dziedziczone duplikacje genów ojcowskich lub izodisomia rodzicielska genów ojcowskich, chociaż coraz częściej rozpoznawane są zaburzenia metylacji (E).[16] Cukrzyca związana z nimi jest typowo rozpoznawana w pierwszym tygodniu życia i ustępuje około 12. tygodnia życia (B).[15] W niemal 50% przypadków cukrzyca wystąpi ponownie w wieku dziecięcym (B).15 Poza powiększeniem języka obserwowanym u 23% chorych nie występują inne objawy niezwiązane z trzustką (B).[15]
Początkowe stężenia glukozy mogą być bardzo duże (zakres 12–57 mmol/l [216–1026 mg/dl – przyp. red.]) i dlatego na początku leczenia stosuje się insulinę, chociaż jej dawkę można szybko zmniejszyć. Z uwagi na występowanie nawrotów pacjenci powinni być poddawani regularnej, corocznej kontroli. W razie ponownego wystąpienia cukrzycy, początkowo nie występuje insulinozależność i chorzy mogą być leczeni dietą, chociaż później często wymagają leczenia insuliną (E).[14] Nieznana jest skuteczność leków doustnych, takich jak pochodne sulfonylomocznika lub metformina.
Utrwalona cukrzyca noworodków, przemijająca cukrzyca noworodków i cukrzyca rozpoznana w pierwszych 6 miesiącach życia spowodowana mutacją genu zależnego od ATP kanału potasowego
Stymulowane przez glukozę wydzielanie insuliny wymaga zamknięcia zależnego od ATP kanału potasowego komórki β. Kanał ten składa się z czterech podjednostek kanału potasowego (Kir6.2) i z czterech podjednostek receptora pochodnych sulfonylomocznika. Mutacja aktywująca genów kodujących te podjednostki powoduje trwałe otwarcie kanału, przeciwdziałając wydzielaniu insuliny, a przez to powodując cukrzycę noworodków. Mutacje Kir6.2 znajdują się na drugim miejscu pod względem częstości wszystkich mutacji stwierdzanych u chorych, u których rozpoznanie cukrzycy postawiono w okresie pierwszych 6 miesięcy życia (B).[17,18] Podczas gdy u niektórych (10%) występuje ulegająca spontanicznej remisji postać cukrzycy, która może w późniejszym okresie nawrócić, to u większości chorych cukrzyca noworodków od początku jest utrwalona (C).[22] U większości chorych cukrzyca ma charakter izolowany, chociaż u 20% stwierdza się objawy neurologiczne. Mimo że chorzy są heterozygotami, u większości nie stwierdza się obciążającego wywiadu rodzinnego, gdyż w 90% przypadków mutacje są spontaniczne.
Najcięższym zaburzeniem jest znaczne opóźnienie rozwoju psychomotorycznego i społecznego oraz uogólniona padaczka, której często towarzyszy hipsarytmia, podobnie jak w zespole Westa (C).[17] Choroba jest określana jako zespół DEND (developmental delay, epilepsy, neonatal diabetes: opóźnienie rozwoju, padaczka, cukrzyca noworodkowa).[18] Częściej występuje częściowy zespół DEND, w którym u chorych stwierdza się mniejszy stopień upośledzenia rozwoju i brak padaczki.[18] Chorzy z mutacją Kir6.2 wykazują wszystkie cechy insulinozależności, u 30% w chwili rozpoznania występuje cukrzycowa kwasica ketonowa i zwykle mają nieoznaczalne stężenie peptydu C, z tego powodu są leczeni insuliną (C).[18] Niedawno wykazano, że chorzy ci nie tylko mogą być skutecznie leczeni doustnymi pochodnymi sulfonylomocznika, ale mogą również osiągnąć lepszą kontrolę glikemii bez nasilenia hipoglikemii. W porównaniu z dorosłymi dawki w przeliczeniu na kilogram masy ciała są duże, a typowa dawka glibenklamidu wynosi 0,5 mg/kg mc./dobę, chociaż niektórzy chorzy mogą wymagać dawki aż 1 mg/kg mc./dobę (C).[23-29] Z czasem u wielu chorych można zmniejszyć dawkę pochodnych sulfonylomocznika, utrzymując bardzo dobrą kontrolę glikemii (E). Mutacje SUR1 są przyczyną 12% przypadków cukrzycy noworodków i często powodują cukrzycę przemijającą.[30,31]
Zespół Wolcott i Rallisona
Zespół Wolcott i Rallisona (WRS) jest rzadkim, dziedziczonym autosomalnie recesywnie zaburzeniem charakteryzującym się cukrzycą o wczesnym początku, dysplazją nasadową, uszkodzeniem nerek, ostrą niewydolnością wątroby i opóźnieniem rozwoju (B).[32,33] Jest związany z mutacją EIF2AK3.[34] Cukrzyca zwykle występuje w okresie niemowlęcym, ale może wystąpić także później i łączy się z niezwiązaną z autoimmunizacją utratą komórek β, prowadzącą do niedoboru insuliny. Wymagane jest leczenie insuliną. Wystąpienie WRS należy brać pod uwagę u każdego chorego z cukrzycą rozpoznaną w pierwszych trzech latach życia, u którego stwierdza się dysplazję nasadową lub ostrą niewydolność wątroby (C,E).[32]
Inne przyczyny cukrzycy noworodków
W tabeli 1 przedstawiono objawy kliniczne cukrzyc noworodków o innej przyczynie. Najczęstszymi innymi przyczynami są mutacje INS, które nie powodują objawów pozatrzustkowych.[35] Najbardziej przydatnymi badaniami poprzedzającymi molekularne badania genetyczne są badania obrazowe trzustki potwierdzające jej obecność oraz rozmiary, ocena zewnątrzwydzielniczej funkcji trzustki i oznaczenie autoprzeciwciał trzustkowych (występujących w zespole IPEX) (E). Wszystkie inne przyczyny cukrzycy noworodków wymagają leczenia insuliną. Niektórzy pediatrzy uważają, że najłatwiej jest leczyć tych chorych podskórnymi wlewami insuliny za pomocą pompy. U chorych z aplazją trzustki konieczna jest suplementacja enzymów trzustkowych.
Cukrzyca występująca rodzinnie
Najczęstsze przyczyny występującej rodzinnie cukrzycy lub hiperglikemii przedstawiono w tabeli 2.
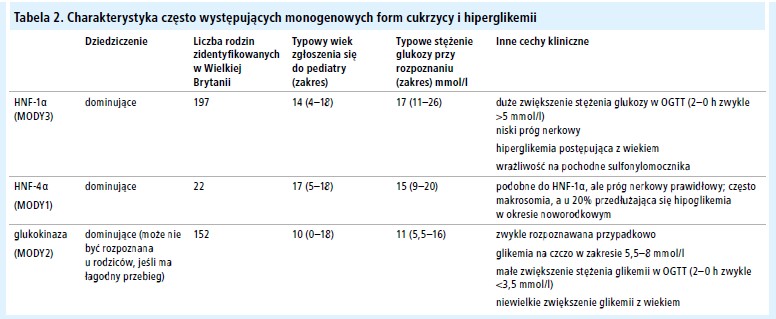
Dzieci i młodzi dorośli z cukrzycą oraz obciążającym wywiadem rodzinnym w kierunku cukrzycy: mutacje genu czynnika jądrowego hepatocyte nuclear factor hepatocytów 1α ( 1 alpha – HNF-1α) (MODY3) Ewentualność cukrzycy monogenowej powinna być brana pod uwagę w każdym przypadku, gdy rodzic pacjenta choruje na cukrzycę, nawet jeśli u pacjenta rozpoznano cukrzycę typu 1 lub 2 (E). Najczęstszą postacią występującej rodzinnie cukrzycy monogenowej (dawniej określaną jako MODY [maturity-onset diabetes of the young]) jest mutacja HNF-1α (B).[36] Cechy kliniczne chorych z mutacją HNF-1α są następujące:
Leczenie. Chorzy z mutacjami genu HNF-1α mogą początkowo być leczeni dietą, chociaż występuje u nich znaczna poposiłkowa hiperglikemia, ponieważ z powodu upośledzenia funkcji komórek β nie dochodzi do zwiększenia wydzielania insuliny po spożyciu pokarmów o dużej zawartości węglowodanów.[37]
Większość chorych wymaga leczenia farmakologicznego, ponieważ z wiekiem wykazują stopniowe pogorszenie kontroli glikemii i są obciążeni ryzykiem powikłań o charakterze mikro- i makroangiopatii (B, C).[38]
Pierwszym lekiem stosowanym u dzieci, u których nie udaje się uzyskać kontroli glikemii za pomocą insulin, powinna być podawana w małej dawce pochodna sulfonylomocznika, która powoduje 4-krotnie silniejsze zmniejszenie glikemii w porównaniu z metforminą (A).[39] U chorych z tym typem cukrzycy występuje szczególna wrażliwość na pochodne sulfonylomocznika i dopóki nie stwierdza się u nich problemów z hipoglikemią, takie leczenie może być u nich stosowane przez wiele dziesięcioleci (C).[34] Kontrola glikemii w trakcie leczenia pochodnymi sulfonylomocznika często jest lepsza od osiąganej w trakcie leczenia insuliną, zwłaszcza u dzieci i młodych dorosłych.[40] Dawka pochodnej sulfonylomocznika początkowo powinna być mała (1/4 typowej dawki początkowej dla dorosłych), aby uniknąć hipoglikemii (E). Jeśli hipoglikemia występuje pomimo dostosowywania dawki pochodnych sulfonylomocznika, takich jak gliklazyd podawany raz lub dwa razy na dobę, można rozważyć zastosowanie preparatu o przedłużonym uwalnianiu albo podawanego do posiłków krótko działającego leku, takiego jak nateglinid (C).[4]
Dzieci i młodzi dorośli z cukrzycą oraz obciążającym wywiadem rodzinnym w kierunku cukrzycy: mutacje genu czynnika jądrowego hepatocytów 4 α (hepatocyte nuclear factor 4 alpha – HNF-4α) (MODY1) Cukrzyca spowodowana mutacjami genu HNF-4. występuje znacznie rzadziej (tab. 2) niż cukrzyca spowodowana mutacjami genu HNF-1α, ale cechuje się podobną charakterystyką, z wyjątkiem niskiego progu nerkowego oraz starszego wieku w momencie rozpoznania (C).[45] Mutacje HNF-4α powinny być brane pod uwagę w przypadku negatywnego wyniku sekwencjonowania HNF-1α, a przy występowaniu objawów klinicznych silnie sugerujących mutacje HNF-1α.[45] U chorych często występuje wrażliwość na pochodne sulfonylomocznika (C).[46]
Inne przyczyny cukrzyc występujących rodzinnie
Opisano kilka rodzin z insulinoniezależną cukrzycą, dziedziczoną autosomalnie dominu jąco, u których występowały mutacje genów IPF1 (MODY4),[47] NeuroD1 (MODY6),[48,49] a ostatnio lipazy karboksylowej (CEL) (MODY7).[50] Są one na tyle rzadkie, że nie ma potrzeby testowania pod ich kątem dzieci chorych na cukrzycę, z wyjątkiem celów naukowych (E) lub gdy występują dodatkowe cechy fenotypowe, takie jak zaburzenia zewnątrzwydzielniczej funkcji trzustki.[50]
Łagodna hiperglikemia na czczo spowodowana mutacjami glukokinazy (MODY2)
U dzieci i młodych dorosłych rzadko stwierdza się zwiększenie stężenia glukozy na czczo do wartości 5,5–8,5 mmol/l (99–144 mg/dl – przyp. red.). Zawsze budzi to podejrzenie, że u tych osób może rozwinąć się cukrzyca typu 1 albo że u chorego występuje cukrzyca typu 2. Tymczasem znaczna część tych chorych z utrwaloną hiperglikemią na czczo jest heterozygotami w zakresie mutacji genu glukokinazy. Cechy fenotypowe związane z mutacjami glukokinazy są bardzo podobne we wszystkich rodzajach mutacji. Wymienione poniżej objawy sugerują rozpoznanie mutacji glukokinazy:
Leczenie. Hiperglikemia na czczo nie ulega znaczącemu pogorszeniu z czasem, a stężenie glukozy jest kontrolowane na poziomie wyższych wartości docelowych.[37] Rzadko wiąże się to z jakimikolwiek powikłaniami o charakterze mikro- i makroangiopatii, nawet jeśli przez całe życie nie stosuje się leczenia (C).[51]
Co ważne, chorzy nie wymagają leczenia w wieku dziecięcym. Odpowiedź na doustne leki hipoglikemizujące lub insulinę jest bardzo mała albo nie występuje wcale (E). Egzogenna insulina powoduje zmniejszenie wydzielania insuliny endogennej i ustabilizowanie glikemii, dlatego stosowanie insuliny u dzieci z tym typem cukrzycy nie powoduje znacznej hipoglikemii.
Zespoły genetyczne związane z cukrzycą
W przypadku gdy cukrzyca u dziecka związana jest z występowaniem innych chorób dotyczących wielu układów, należy rozważyć możliwość występowania zespołu monogenowego.
W ocenie cech klinicznych oraz w uzyskaniu informacji, czy odpowiedzialny za zaburzenie gen został zidentyfikowany oraz czy możliwe jest przeprowadzenie badań genetycznych, pomocna może być strona internetowa OMIM (Online Mendelian inheritance in Man) (dostęp przez stronę NCBI www.ncbi.nlm.nih.gov/entrez/query.fcgi). W przypadkach opisanych i jeszcze nieopisanych zespołów można uzyskać pomoc poprzez bazę rzadkich typów cukrzycy ISPAD (ISPAD rare diabetes collection; kontakt przez link na stronie internetowej ISPAD lub przez www.diabetesgenes.org). Najczęściej występujące zespoły genetyczne związane z cukrzycą wymieniono poniżej.
Moczówka prosta, cukrzyca, zanik nerwu wzrokowego, głuchota: zespół DIDMOAD
DIDMOAD – diabetes insipidus, diabetes mellitus, optic atrophy, deafness; zespół Wolframa Zespół Wolframa jest dziedziczonym autosomalnie recesywnie zespołem, w którym podstawą rozpoznania jest współistnienie cukrzycy i postępującego zaniku nerwu wzrokowego przed ukończeniem 16. roku życia.[52] Zespół częściej występuje w grupach etnicznych, w których małżeństwa zawierane są przez spokrewnione osoby. Do pozostałych objawów należą: obustronna głuchota czuciowo-odbiorcza, moczówka prosta, poszerzenie układu kielichowo-miedniczkowego, ataksja osiowa oraz inne różnorodne objawy neurologiczne, z kompletnym fenotypem stwierdzanym u 75% chorych z częstością zwiększającą się z wiekiem. Kolejność pojawiania się objawów neurologicznych może być różna nawet w obrębie jednej rodziny. Średni wiek zgonu w zespole Wolframa to 30 lat.[52] Mutacje w obrębie genu zespołu Wolframa (WS1) występują u co najmniej 90% chorych z objawami klinicznymi tego zespołu.[53-55]
Cukrzyca nie jest spowodowana autoimmunizacją, a niedobór insuliny występuje średnio w 6. roku życia.[52] Chorzy wymagają leczenia insuliną od momentu rozpoznania, ale nie stwierdza się obecności autoprzeciwciał (C).[52]
Niedokrwistość megaloblastyczna odpowiadająca na leczenia tiaminą (zespół Rogera)
Niedokrwistość megaloblastyczna odpowiadająca na leczenia tiaminą (thiamine responsive megaloblastic anaemia – TRMA) jest rzadkim, dziedziczonym recesywnie zespołem genetycznym, w którym występuje niedokrwistość megaloblastyczna o wczesnym początku (odpowiadająca na leczenie tiaminą) związana z cukrzycą i głuchotą czuciowo-odbiorczą.
Jest ona wynikiem mutacji genu SLC19A2.[56] Cukrzyca, która z natury jest związana z niedoborem insuliny, u niektórych chorych odpowiada na leczenie tiaminą, chociaż w dłuższym okresie wydaje się jednak rozwijać zapotrzebowanie na insulinę (C).[57] Głuchota nie odpowiada na leczenie tiaminą.
Wrodzona torbielowatość nerek i cukrzyca spowodowana mutacją czynnika jądrowego hepatocytów 1β (hepatic nuclear factor 1-β – HNF-1β)
Chociaż dawniej zespół ten zaliczano do podgrupy cukrzyc występujących rodzinnie (MODY5), obecnie wiadomo, że u chorych z mutacją HNF-1β izolowana cukrzyca występuje rzadko.[58] Zaburzenia rozwojowe nerek, zwłaszcza torbiele nerek i dysplazja nerek występujące u niemal wszystkich chorych z mutacją lub delecją genu,[59] mogą być rozpoznane wewnątrzmacicznie i poprzedzać rozpoznanie cukrzycy (B). Do innych objawów, które mogą wystąpić u dzieci, należą: zaburzenia rozwojowe macicy i zewnętrznych narządów płciowych, hiperurykemia, dna moczanowa i nieprawidłowa aktywność enzymów wątrobowych.[58] Rozpoznanie mutacji HNF-1β powinno być brane pod uwagę u każdego dziecka chorego na cukrzycę, u którego występuje też niezwiązana z cukrzycą choroba nerek.
Chorzy z mutacjami HNF-1β, w przeciwieństwie do chorych z mutacjami HNF-1β, nie są wrażliwi na pochodne sulfonylomocznika i dlatego zwykle wymagają leczenia insuliną.[60] Trzustka ulega zmniejszeniu, które obejmuje zarówno część wewnątrzwydzielniczą, jak i zewnątrzwydzielniczą, a u większości chorych występuje subkliniczna niewydolność zewnątrzwydzielniczej funkcji trzustki,[61] ale nie jest oczywiste, czy w przypadku braku objawów powinna być leczona.
Cukrzyce dziedziczone mitochondrialnie
Przekazanie matczynego DNA mitochondrialnego (mtDNA) z mutacją lub delecją może spowodować cukrzycę dziedziczoną od matki, chociaż nieczęsto ujawnia się ona w wieku dziecięcym. Wprawdzie wskazywano na kilka mutacji i delecji, ale najlepiej udokumentowano związek z pojedynczą zamianą nukleotydu w pozycji 3243 (A na G) w mitochondrialnym tRNA (leu[UUR]) (B).[62] Ta sama mutacja występuje w zespole MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, stroke-like syndrome; miopatia mitochondrialna, encefalopatia, kwasica mleczanowa i rzekomy udar), a pewnego stopnia nakładanie tych zespołów może występować u członków tej samej rodziny. Cukrzyce dziedziczone mitochondrialnie często są związane z głuchotą czuciowo-odbiorczą i niskim wzrostem. Cukrzyca charakteryzuje się postępującą, niezwiązaną z autoimmunizacją niewydolnością komórek β i może szybko prowadzić do konieczności stosowania insuliny.
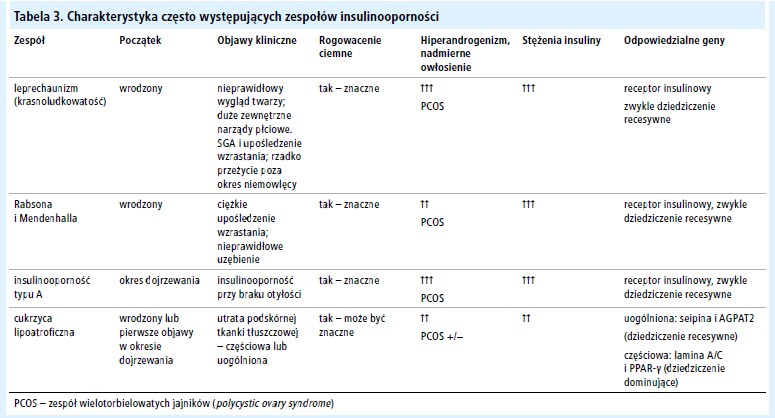
Zespoły insulinooporności: insulinooporność typu A (leprechaunism, "krasnoludkowatość"), zespół Rabsona i Mendenhalla i cukrzyca lipoatroficzna
Kluczowymi cechami wszystkich zespołów insulinooporności są: rogowacenie ciemne, nadmiar androgenów oraz bardzo znacznie zwiększone stężenie insuliny przy braku otyłości.[1] Im cięższa insulinooporność i wcześniejszy początek objawów, tym bardziej prawdopodobne jest wystąpienie cukrzycy (C).1 Zestawienie niektórych kluczowych objawów klinicznych zamieszczono poniżej (adaptowano na podstawie Musso i wsp.[1]).
Leczenie ciężkiej insulinooporności jest bardzo trudne, a u większości chorych kontrola glikemii jest niedostateczna i często występują odległe powikłania (C).[1] Leczenie polega na podawaniu leków zwiększających wrażliwość na insulinę – metforminy i pochodnych tiazolidynedionu, ale ich skuteczność w przypadku bardzo ciężkiej insulinooporności jest ograniczona. Główną metodą leczenia jest stosowanie insuliny, często wymagane jest użycie insuliny w stężeniu 500 j.m./ml (U500) i osobistych pomp insulinowych.[1] W częściowej lipodystrofii korzystne może być stosowanie metforminy, a insulina nie jest wymagana we wczesnym stadium choroby (C).[63] W uogólnionej lipodystrofii wynik leczenia cukrzycy za pomocą rekombinowanej leptyny[64] może być bardzo dobry, ale tego rodzaju terapia dostępna jest tylko w ramach prowadzonych badań naukowych.
Zalecenia
Postępy genetyki molekularnej doprowadziły do zidentyfikowana genów związanych z większością scharakteryzowanych klinicznie podtypów cukrzycy. Zidentyfikowanie genów wyjaśniło zmienność przebiegu klinicznego chorób definiowanych na podstawie czasu ich wystąpienia, np. cukrzyca noworodków, MODY. Obecnie metody genetyki molekularnej są stosowane jako badanie diagnostyczne, które może pomóc w określeniu rozpoznania i leczenia u dzieci chorych na cukrzycę. Z uwagi na duży koszt tych badań powinno się je wykonywać w tych przypadkach, w których na podstawie przebiegu klinicznego spodziewany jest pozytywny wynik (E).
Piśmiennictwo
1. Musso C., Cochran E., Moran S.A., Skarulis M.C., Oral E.A., Taylor S., et al.: Clinical course of genetic diseases of the insulin receptor (type A and Rabson-Mendenhall syndromes): a 30-year prospective. Medicine (Baltimore), 2004; 83 (4): 209–222
2. Moller A.M., Dalgaard L.T., Pociot F., Nerup J., Hansen T., Pedersen O.: Mutations in the hepatocyte nuclear factor-1alpha gene in Caucasian families originally classified as having Type Idiabetes. Diabetologia, 1998; 41: 1528–1531
3. Lambert A.P., Ellard S., Allen L.I., Gallen I.W., Gillespie K.M., Bingley P.J., et al.: Identifying hepatic nuclear factor 1alpha mutations in children and young adults with a clinical diagnosis of type 1 diabetes. Diabetes Care, 2003; 26 (2): 333–337
4. Iafusco D., Stazi M.A., Cotichini R., Cotellessa M., Martinucci M.E., Mazzella M., et al.: Permanent diabetes mellitus in the first year of life. Diabetologia, 2002; 45 (6): 798–804
5. Tillil H., Kobberling J.: Age-corrected empirical genetic risk estimates for first-degree relatives of IDDM patients. Diabetes, 1987; 36 (1): 93–99
6. Hathout E.H., Sharkey J., Racine M., Thomas W ., Nahab F., El Shahawy M., et al.: Diabetic autoimmunity in infants and pre-schoolers with type 1 diabetes. Pediatr. Diabetes, 2000; 1 (3): 131–134
7. Borg H., Marcus C., Sjoblad S., Fernlund P., Sundkvist G.: Insulin autoantibodies are of less value compared with islet antibodies in the clinical diagnosis of autoimmune type 1 diabetes in children older than 3 yr of age. Pediatr. Diabetes, 2002; 3 (3): 149–154
8. Sabbah E., Savola K., Kulmala P., Veijola R., Vahasalo P., Karjalainen J., et al.: Diabetes-associated autoantibodies in relation to clinical characteristics and natural course in children with newly diagnosed type 1 diabetes. The Childhood Diabetes In Finland Study Group. J. Clin. Endocrinol. Metab., 1999; 84 (5): 1534–1539
9. Gungor N., Hannon T., Libman I., Bacha F., Arslanian S.: Type 2 diabetes mellitus in youth: the complete picture to date. Pediatr. Clin. North Am., 2005; 52 (6): 1579–1609
10. Ehtisham S., Barrett T.G., Shaw N.J.: Type 2 diabetes mellitus in UK children–an emerging problem. Diabet. Med., 2000; 17 (12): 867–871
11. Ehtisham S., Hattersley A.T., Dunger D.B., Barrett T.G.: First UK survey of paediatric type 2 diabetes and MODY. Arch. Dis. Child., 2004; 89 (6): 526–529
12. American Diabetes Association. Type 2 diabetes in Children and Adolescents. Diabetes Care, 2000; 23: 381–389
13. Slingerland A.S., Hattersley A.T.: Mutations in the Kir6.2 subunit of the KATP channel and permanent neonatal diabetes: new insights and new treatment. Ann. Med., 2005; 37 (3): 186–195
14. Polak M., Shield J.: Neonatal and very-early-onset diabetes mellitus. Semin. Neonatol., 2004; 9 (1): 59–65
15. Temple I.K., Gardner R.J., Mackay D.J., Barber J.C., Robinson D.O., Shield J.P.: Transient neonatal diabetes: widening the understanding of the etiopathogenesis of diabetes. Diabetes, 2000; 49 (8): 1359–1366
16. Gardner R.J., Mackay D.J., Mungall A.J., Polychronakos C., Siebert R., Shield J.P., et al.: An imprinted locus associated with transient neonatal diabetes mellitus. Hum. Mol. Genet., 2000; 9 (4): 589–596
17. Gloyn A.L., Pearson E.R., Antcliff J.F., Proks P., Bruining G.J., Slingerland A.S., et al.: Activating mutations in the gene encoding the ATP-sensitive potassium channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med., 2004; 350 (18): 1838–1849
18. Hattersley A.T., Ashcroft F.M.: Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes, 2005; 54 (9): 2503–2513
19. Njolstad P., Sovik O., Cuesta-Munoz A., Bjorkhaug L., Massa O., Barbetti F., et al.: Neonatal diabetes mellitus due to complete glucokinase deficiency. N. Engl. J. Med., 2001; 344: 1588–1592
20. Njolstad P.R., Sagen J.V., Bjorkhaug L., Odili S., Shehadeh N., Bakry D., et al.: Permanent neonatal diabetes mellitus due to glucokinase deficiency- an inborn error of glucose-insulin signalling pathway. Diabetes, 2003; 52 (11): 2854–2860
21. Mackay D.J., Coupe A.M., Shield J.P., Storr J.N., Temple I.K., Robinson D.O.: Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum. Genet., 2002; 110 (2): 139–144
22. Gloyn A.L., Reimann F., Girard C., Edghill E.L., Proks P., Pearson E.R., et al.: Relapsing diabetes can result from moderately activating mutations in KCNJ11. Hum. Mol. Genet., 2005; 14 (7): 925–934
23. Inagaki N., Gonoi T., Clement J.P. 4th, et al.: Reconstitution of IKATP: an inward rectifier subunit plus the sulphonylurea receptor. Science, 1995; 270: 1166–1170
24. Sagen J.V., Raeder H., Hathout E., Shehadeh N., Gudmundsson K., Baevre H., et al.: Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes, 2004; 53 (10): 2713–2718
25. Codner E., Flanagan S., Ellard S., Garcia H., Hattersley A.T.: High-dose glibenclamide can replace insulin therapy despite transitory diarrhea in early-onset diabetes caused by a novel R201L Kir6.2 mutation. Diabetes Care, 2005; 28 (3): 758–759
26. Massa O., Iafusco D., D'amato E., Gloyn A.L., Hattersley A.T., Pasquino B., et al.: KCNJ11 activating mutations in Italian patients with permanent neonatal diabetes. Hum. Mutat., 2005; 25 (1): 22–27
27. Hattersley A.T.: Molecular genetics goes to the diabetes clinic. Clin. Med., 2005; 5 (5): 476–481
28. Zung A., Glaser B., Nimri R., Zadik Z.: Glibenclamide Treatment in Permanent Neonatal Diabetes Mellitus due to an Activating Mutation in Kir6.2. J. Clin. Endocrinol. Metab., 2004; 89 (11): 5504–5507
29. Pearson E.R., Flechtner I., Njolstad P.R., Malecki M.T., Flanagan S.E., Larkin B., Ashcroft F.M., Klimes I., Codner E., Iotova V., Slingerland A.S., Shield J., Robert J.J., Holst J.J., Clark P.M., Ellard S., Sovik O., Polak M., Hattersley A.T.: Neonatal Diabetes International Collaborative Group. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N. Engl. J. Med., 2006; 355 (5): 467–477
30. Proks P., Arnold A.L., Bruining J.,Girard C., Flanagan S.E., Larkin B., Colclough K., Hattersley A.T., Ashcroft F.M., Ellard S.: A heterozygous activating mutation in the sulphonylurea receptor SUR1 (ABCC8) causes neonatal diabetes. Hum. Mol. Genet., 2006; 15 (11): 1793–1800
31. Babenko A.P., Polak M., Cave H., Busiah K., Czernichow P., Scharfmann R., Bryan J., Aguilarbryan L., Vaxillaire M., Froguel P.: Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med., 2006; 355 (5): 456–466
32. Iyer S., Korada M., Rainbow L., Kirk J., Brown R.M., Shaw N., et al.: Wolcott-Rallison syndrome: a clinical and genetic study of three children, novel mutation in EIF2AK3 and a review of the literature. Acta Paediatr., 2004; 93 (9): 1195–1201
33. Senee V., Vattem K.M., Delepine M., Rainbow L.A., Haton C., Lecoq A., et al.: Wolcott-Rallison Syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes, 2004; 53 (7): 1876–1883
34. Delepine M., Nicolino M., Barrett T., Golamaully M., Lathrop G.M., Julier C.: EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet., 2000; 25 (4): 406–409
35. Stoy J., Edghill E.L., Flanagan S.E., Ye H., Paz V.P., Pluzhnikov A., et al.: Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. USA, 2007; 104: 15 040–15 044
36. Stride A., Hattersley A.T.: Different genes, different diabetes: lessons from maturity-onset diabetes of the young. Ann. Med., 2002; 34 (3): 207–216
37. Stride A., Vaxillaire M., Tuomi T., Barbetti F., Njolstad P.R., Hansen T., et al.: The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia, 2002; 45 (3): 427–435
38. Pearson E.R., Liddell W .G., Shepherd M., Corrall R.J., Hattersley A.T.: Sensitivity to sulphonylureas in patients with hepatocyte nuclear factor-1alpha gene mutations: evidence for pharmacogenetics in diabetes. Diabet. Med., 2000; 17: 543–545
39. Pearson E.R., Starkey B.J., Powell R.J., Gribble F.M., Clark P.M., Hattersley A.T.: Genetic aetiology of hyperglycaemia determines response to treatment in diabetes. Lancet, 2003; 362 (9392): 1275–1281
40. Byrne M.M., Sturis J., Menzel S., Yamagata K., Fajans S.S., Dronsfield M.J., et al.: Altered insulin secretory responses to glucose in diabetic and nondiabetic subjects with mutations in the diabetes susceptibility gene MODY3 on Chromosome 12. Diabetes, 1996; 45: 1503–1510
41. Isomaa B., Henricsson M., Lehto M., Forsblom C., Karanko S., Sarelin L., et al.: Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia, 1998; 41 (4): 467–473
42. Pearson E.R., Starkey B.J., Powell R.J., Gribble F.M., Clark P.M., Hattersley A.T.: Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet, 2003; 362 (9392): 1275–1281
43. Shepherd M., Pearson E.R., Houghton J., Salt G., Ellard S., Hattersley A.T.: No deterioration in glycemic control in HNF-1alpha maturity-onset diabetes of the young following transfer from long-term insulin to sulphonylureas. Diabetes Care, 2003; 26 (11): 3191–3192
44. Tuomi T., Honkanen E.H., Isomaa B., Sarelin L., Groop L.C.: Improved prandial glucose control with lower risk of hypoglycemia with nateglinide than with glibenclamide in patients with maturity-onset diabetes of the young type 3. Diabetes Care, 2006; 29 (2): 189–194
45. Pearson E.R, Pruhova S., Tack C.J., Johansen A., Castleden H.A., Lumb P.J., et al.: Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia, 2005; 48 (5): 878–885
46. Fajans S.S., Brown M.B.: Administration of sulfonylureas can increase glucose-induced insulin secretion for decades in patients with maturity-onset diabetes of the young. Diabetes Care, 1993; 16 (9): 1254–1261
47. Stoffers D.A., Ferrer J., Clarke W .L., Habener J.F.: Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat. Genet., 1997; 17: 138–139
48. Malecki M.T., Jhala U.S., Antonellis A., Fields L., Doria A., Orban T., et al.: Mutations in NEUROD1 are associated with the development of Type 2 diabetes mellitus. Nat. Genet., 1999; 23 (3): 323–328
49. Kristinsson S.Y., Thorolfsdottir E.T., Talseth B., Steingrimsson E., Thorsson A.V., Helgason T., et al.: MODY in Iceland is associated with mutations in HNF- 1alpha and a novel mutation in NeuroD1. Diabetologia, 2001; 44 (11): 2098–2103
50. Raeder H., Johansson S., Holm P.I., Haldorsen I.S., Mas E., Sbarra V., et al.: Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat. Genet., 2006; 38 (1): 54–62
51. Velho G., Blanché H., Vaxillaire M., Bellanné-Chantelot C., Pardini V.C., Timsit J., et al.: Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia, 1997; 40: 217–224
52. Barrett T.G., Bundey S.E., Macleod A.F.: Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet, 1995; 346: 1458–1463
53. Strom T.M., Hortnagel K., Hofmann S., Gekeler F., Scharfe C., Rabl W ., et al.: Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum. Mol. Genet., 1998; 7 (13): 2021–2028
54. Inoue H., Tanizawa Y., Wasson J., Behn P., Kalidas K., Bernal-Mizrachi E., et al.: A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat. Genet., 1998; 20 (2): 143–148
55. Hardy C., Khanim F., Torres R., Scott-Brown M., Seller A., Poulton J., et al.: Clinical and molecular genetic analysis of 19 Wolfram syndrome kindreds demonstrating a wide spectrum of mutations in WFS1. Am. J. Hum. Genet., 1999; 65 (5): 1279–1290
56. Labay V., Raz T., Baron D., Mandel H., Williams H., Barrett T., et al.: Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat. Genet., 1999; 22 (3): 300–304
57. Ozdemir M.A., Akcakus M., Kurtoglu S., Gunes T., Torun Y.A.: TRMA syndrome (thiamine-responsive megaloblastic anemia): a case report and review of the literature. Pediatr. Diabetes, 2002; 3 (4): 205–209
58. Bingham C., Hattersley A.T.: Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol. Dial. Transplant., 2004; 19 (11): 2703–2708
59. Bellanné-Chantelot C., Clauin S., Chauveau D., Collin P., Daumont M., Douillard C., et al.: Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes, 2005; 54 (11): 3126–3132
60. Pearson E.R., Badman M.K., Lockwood C.R., Clark P.M., Ellard S. Bingham C., et al.: Contrasting diabetes phenotypes associated with hepatocyte nuclear factor-1alpha and -1beta mutations. Diabetes Care, 2004; 27 (5): 1102–1107
61. Bellanné-Chantelot C., Chauveau D., Gautier J.F., Dubois-Laforgue D., Clauin S., Beaufils S., et al.: Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann. Intern. Med., 2004; 140 (7): 510–517
62. Van Den Ouweland J.M., Lemkes H.H., Ruitenbeek W ., Sandkuijl L.A., De Vijlder M.F., Struyvenberg P.A., et al.: Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet., 1992; 1 (5): 368–371
63. Owen K.R., Donohoe M., Ellard S., Hattersley A.T.: Response to treatment with rosiglitazone in familial partial lipodystrophy due to a mutation in the LMNA gene. Diabet. Med., 2003; 20 (10): 823–827
64. Petersen K.F., Oral E.A., Dufour S., Befroy D., Ariyan C., Yu C., et al.: Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J. Clin. Invest., 2002; 09 (10): 1345–1350
Konflikt interesów
Autorzy nie zgłosili konfliktu interesów.