Tłumaczyła dr n. med. Katarzyna Pawińska-Wąsikowska
Konsultowała prof. dr hab. n. med. Przemysława Jarosz-Chobot, Klinika Diabetologii Dziecięcej
Śląskiego Uniwersytetu Medycznego w Katowicach
Skróty: ADA – American Diabetes Association, CFRD – cukrzyca wtórna do mukowiscydozy, CGM – ciągłe monitorowanie glikemii, IGT – nieprawidłowa tolerancja glukozy, OGTT – doustny test obciążenia glukozą
Podsumowanie i zalecenia
- Cukrzyca wtórna do mukowiscydozy (cystic fibrosis-related diabetes – CFRD) stanowi najczęstszą chorobę występującą w przebiegu mukowiscydozy.
- CFRD jest chorobą o złożonej patogenezie, spowodowaną zniszczeniem komórek wysp trzustki z jednoczesnym niedoborem insuliny i glukagonu, prowadzącym do zmiennej oporności na insulinę, konieczności stosowania diety wysokokalorycznej oraz zaburzeń funkcji przewodu pokarmowego, w tym opóźnionego opróżniania żołądka, zaburzeń motoryki jelit oraz do chorób wątroby.
- CFRD może wystąpić w każdym wieku, również u niemowląt, a częstość jej występowania zwiększa się wraz z wiekiem.
- U nielicznych chorych na mukowiscydozę tolerancja glukozy jest prawidłowa, ale nawet jeśli glikemia na czczo lub 2 h po doustnym obciążeniu glukozą (oral glucose tolerance test – OGTT) jest w normie, zmienną, przemijającą hiperglikemię poposiłkową można często wykryć dzięki ciągłemu monitorowaniu glikemii (continuous glucose monitoring – CGM).
- U dzieci chorych na mukowiscydozę wraz z wiekiem dochodzi do postępującego upośledzenia tolerancji glukozy, początkowo pod postacią nieokreślonej hiperglikemii, następnie nieprawidłowej tolerancji glukozy (impaired glucose tolerance – IGT), a w końcu cukrzycy.
- Wczesny etap CFRD charakteryzuje się prawidłowym stężeniem glukozy na czczo, z czasem jednak pojawia się hiperglikemia na czczo. Stężenie glukozy oznaczane w różnych punktach czasowych może być zmienne, zależne od aktualnego stanu układu oddechowego oraz występowania zakażeń.
- Objawy CFRD mogą się rozwijać skrycie, jednak większość pacjentów nie prezentuje żadnych objawów choroby w momencie rozpoznania. Do manifestacji objawów CFRD dochodzi zwykle w czasie zwiększonej oporności na insulinę (np. zakażenie układu oddechowego, leczenie glikokortykosteroidami [GKS]).
- Rzadko pierwszą manifestacją CFRD jest kwasica ketonowa.
- Za początek CFRD uznaje się datę, kiedy chory na mukowiscydozę spełnił kryteria diagnostyczne cukrzycy, mimo normalizacji glikemii w późniejszych oznaczeniach (E).
- U pacjenta z mukowiscydozą w stabilnej fazie choroby rozpoznanie CFRD należy ustalić na podstawie standardowych kryteriów American Diabetes Association (ADA) (E).
| Tabela 1. Porównanie cukrzycy typu 1, typu 2 i cukrzycy wtórnej do mukowiscydozy | |||
|---|---|---|---|
| Cukrzyca typu 1 | Cukrzyca typu 2 | CFRD | |
| częstość | 0,2% | 11% | 35% |
| początek | zwykle ostry | utajony | utajony |
| wiek, w którym najczęściej występują pierwsze objawy | dzieci i młodzież | dorośli | 18–24 lat |
| budowa konstytucjonalna | prawidłowa | otyłość | prawidłowa lub niedobór masy ciała |
| obecność autoprzeciwciał | tak | nie | nie |
| niedobór insuliny | prawie całkowity | częściowy, zmienny | znacznie zmniejszony, ale obecny |
| wrażliwość na insulinę | w pewnym stopniu zmniejszona | poważnie zmniejszona | w pewnym stopniu zmniejszonaa |
| związki ketonowe | tak | rzadko | rzadko |
| typowe leczenie | insulina | dieta, leki doustne, insulina | insulina |
| powikłania o charakterze mikronaczyniowym | tak | tak | tak |
| powikłania o charakterze makronaczyniowym | tak | tak | nie |
| zespół metaboliczny | nie | tak | nie |
| przyczyna zgonu |
choroby układu sercowo-naczyniowego |
choroby układu sercowo-naczyniowego | choroby płuc |
| aWrażliwość na insulinę zmniejsza się znacznie podczas chorób towarzyszących o ostrym przebiegu.
CFRD – cukrzyca wtórna do mukowiscydozy |
|||
- Do rozpoznania CFRD u chorych na mukowiscydozę i towarzyszącą chorobę o ostrym przebiegu konieczne jest stwierdzenie glikemii na czczo ≥126 mg/dl (7,0 mmol/l) lub glikemii 2 h po posiłku ≥200 mg/dl (11,1 mmol/l) utrzymujących się przez ponad 48 h (E).
- U pacjentek chorych na mukowiscydozę i cukrzycę ciążową nie rozpoznaje się CFRD, zaleca się jednak przeprowadzenie badań kontrolnych 6–12 tygodni po porodzie (E).
- Nie jest konieczne rozróżnienie między CFRD z hiperglikemią na czczo oraz bez hiperglikemii na czczo (B).
- Nie zaleca się oznaczania hemoglobiny A1c (HbA1c) jako badania przesiewowego w kierunku CFRD (B).
- Zalecanym badaniem przesiewowym jest OGTT z pomiarem stężenia glukozy 2 h po doustnym podaniu 75 g glukozy (1,75 g/kg mc. [E]).
- Wykonywanie corocznych badań przesiewowych w kierunku CFRD należy rozpocząć przed ukończeniem 10. roku życia u wszystkich chorych na mukowiscydozę, u których dotychczas nie rozpoznano CFRD (B).
- Raz na 3 miesiące pacjentów z CFRD powinien kontrolować wykwalifikowany, wielospecjalistyczny zespół z doświadczeniem w leczeniu mukowiscydozy i cukrzycy (E).
- Pacjentów z CFRD należy objąć ciągłym programem edukacyjnym w zakresie samokontroli cukrzycy, spełniającym krajowe standardy (E).
- W leczeniu CFRD należy stosować insulinę (A).
- Doustne leki przeciwcukrzycowe nie są tak skuteczne jak insulina w poprawie stanu odżywienia i stanu metabolicznego chorych z CFRD, a ich stosowanie należy ograniczyć jedynie do badań naukowych (A).
- Chorzy na CFRD leczeni insuliną powinni samodzielnie kontrolować glikemię przynajmniej 3 razy dziennie (E).
- U chorych na CFRD należy dążyć do osiągnięcia stężeń glukozy określonych w wytycznych ADA dla wszystkich chorych na cukrzycę, zakładając potrzebę indywidualizacji terapii, bowiem u niektórych pacjentów wartości docelowe mogą być mniejsze lub większe niż ustalone w wytycznych (E).
- W ramach monitorowania leczenia insuliną zaleca się wykonywanie oznaczenia HbA1c co 3 miesiące (E).
- Wytyczne żywieniowe dla chorych na mukowiscydozę opracowane przez Cystic Fibrosis Foundation na podstawie wiarygodnych i aktualnych danych naukowych zaleca się również dla chorych na CFRD (E).
- Zaleca się edukację chorych na CFRD i ich opiekunów/ rodziców w zakresie objawów, profilaktyki i leczenia hipoglikemii, w tym stosowania glukagonu (E).
- Zaleca się, aby po upływie 5 lat od rozpoznania CFRD przeprowadzać coroczne badania w kierunku mikroangiopatii cukrzycowej. Jeśli nie wiadomo, kiedy dokładnie rozpoznano CFRD, badania te należy rozpocząć w momencie stwierdzenia po raz pierwszy hiperglikemii na czczo (E).
- Pacjentów z CFRD i nadciśnieniem tętniczym lub mikroangiopatią należy leczyć w standardowy sposób, przy czym nie należy ograniczać spożycia sodu oraz, na ogół, także białka (E).
- U chorych z CFRD i niewydolnością zewnątrzwydzielniczą trzustki zaleca się ocenę profilu lipidowego co roku (E).
Mukowiscydoza jest najczęstszą chorobą genetyczną u osób rasy białej prowadzącą do zgonu, dziedziczoną autosomalnie recesywnie. Mukowiscydoza występuje z częstością 1 na 2500 żywo urodzonych dzieci. Cukrzyca stanowi natomiast najczęstszą chorobę współistniejącą z mukowiscydozą. Istnieją istotne patofizjologiczne różnice między CFRD a cukrzycą zarówno typu 1, jak i 2, dlatego zarówno diagnostyka, jak i leczenie bezwzględnie wymagają szczególnego podejścia (tab. 1.). Do typowych dla mukowiscydozy czynników wpływających na metabolizm glukozy należą: całkowita utrata komórek wysp prowadząca do niedoboru zarówno insuliny, jak i glukagonu; przewlekłe i ostre zakażenia oraz zapalenia układu oddechowego, którym towarzyszą wahania oporności na insulinę; duże zapotrzebowanie kaloryczne związane ze zwiększonym wydatkiem energetycznym i upośledzonym wchłanianiem; ryzyko zagrażającego życiu niedożywienia oraz zaburzenia ze strony przewodu pokarmowego (upośledzone opróżnianie żołądkowe, zaburzenia motoryki jelit i choroby wątroby).
Kryteria diagnostyczne CFRD i nieprawidłowej tolerancji glukozy
W 2010 roku CFRD Guidelines Committe
we współpracy z ADA oraz Cystic Fibrosis Foundation
zmodyfikował kryteria rozpoznania CFRD
dla Ameryki Północnej, przyjęte również przez Pediatric Endrocrine Society.1 Ustalone kryteria
są identyczne z kryteriami opracowanymi dla innych
postaci cukrzycy, ponadto uwzględniają przyjęte
ostatnio oznaczenie HbA1c jako kryterium
diagnostyczne. Należy przy tej okazji podkreślić,
że małe lub prawidłowe wartości HbA1c nie wykluczają
rozpoznania CFRD, bowiem u chorych
na mukowiscydozę wynik oznaczenia HbA1c jest
często fałszywie zaniżony.
cCFRD stanowi część pełnego spektrum postępujących
zaburzeń tolerancji glukozy stwierdzanych w standardowym OGTT (tab. 2.). U nielicznych
chorych na mukowiscydozę stwierdza się prawidłową
tolerancję glukozy (normal glucose tolerance
– NGT). Nawet jeśli glikemia na czczo lub 2 h
po OGTT jest prawidłowa, zmienną, przemijającą
hiperglikemię poposiłkową można często wykryć w warunkach domowych dzięki CGM.2,3 Wraz z pogarszaniem się tolerancji glukozy u chorych
rozwija się nieokreślona hiperglikemia (INDET,
mid-OGTTT; stężenie glukozy >11,1 mmol/l w OGTT w innym punkcie czasowym niż po 2 h
[200 mg/dl]), która następnie poprzez fazę IGT
ostatecznie przechodzi w cukrzycę. Wczesna faza
cukrzycy charakteryzuje się prawidłowym stężeniem
glukozy na czczo, jednak w miarę upływu
czasu pojawia się hiperglikemia w badaniu wykonywanym
na czczo. U chorych na mukowiscydozę
niekiedy obserwuje się izolowaną nieprawidłową
glikemię na czczo (IFG).4,5
U chorych na mukowiscydozę wraz z wiekiem
dochodzi do stopniowego pogarszania tolerancji
glukozy. Jednak stężenie glukozy może się
zmieniać w dowolnym czasie, zależnie od stanu
czynnościowego
układu oddechowego oraz występowania
zakażeń. Zgodnie z wytycznymi CFRD
Guidelines Committee cukrzycę wtórną do mukowiscydozy
rozpoznaje się, gdy pacjent po raz
pierwszy spełnia kryteria rozpoznania cukrzycy,
nawet mimo normalizacji tolerancji glukozy w późniejszych
badaniach. Stanowisko takie przyjęto z uwagi na to, że odległe skutki powikłań mikronaczyniowych i umieralność zależą od czasu trwania
cukrzycy, w tym również wczesnego rozwoju
choroby z okresami zaostrzeń i poprawy tolerancji
glukozy. Gdy u chorego stwierdza się istotną hiperglikemię,
nawet tylko w czasie towarzyszącej
choroby o ostrym przebiegu, z reguły rozwija się
później cukrzyca.1 Hiperglikemię często stwierdza
się u kobiet chorych na mukowiscydozą w okresie
ciąży, z uwagi na występujący jednocześnie
niedobór insuliny.6,7 U kobiet, u których w czasie
ciąży wystąpiła cukrzyca, a które przed ciążą ani
po porodzie nie spełniają kryteriów rozpoznania
cukrzycy, nie rozpoznaje się CFRD.
| Tabela 2. Klasyfikacja nieprawidłowej tolerancji glukozy u chorych na mukowiscydozę | |||
|---|---|---|---|
| Klasyfikacja | Glikemia na czczo (mmol/l)a | Stężenie glukozy po 2 ha | Uwagia |
| prawidłowa (NGT) | <7,0 | <7,8 | wszystkie pomiary glikemii <11,1 |
| nieokreślona (INDET) | <7,0 | <7,8 | glikemia w OGTTw punktach czasowych między 0 a 2 h ≥11,1 |
| nieprawidłowa (IGT) | <7,0 | 7,8–11,1 | |
| CFRD FH (–) | <7,0 | ≥11,1 | |
| CFRD FH (+) | ≥7,0 | ||
| a odpowiednie wartości glikemii w mg/dl: 7,0 mml/l = 126 mg/dl, 7,8 mmol/l = 140 mg/dl,11,1 mml/l = 200 mg/dl
CFRD – cukrzyca wtórna do mukowiscydozy, FH – hiperglikemia na czczo, OGTT – doustny testobciążenia glukozą |
|||
Zapadalność i chorobowość
Zapadalność i częstość występowania cukrzycy u chorych na mukowiscydozę jest większa w porównaniu z odpowiednią grupą wiekową zdrowej
populacji. Częstość występowania CFRD zwiększa
się wraz z wiekiem i w doniesieniach z Danii
wynosiła 4–9% na rok.8 Według danych udostępnionych
przez University of Minnesota zapadalność
na CFRD wynosiła 2,7/100 chorych/rok.9 W ośrodkach, w których powszechnie nie wykonuje
się przesiewowo OGTT, występowanie CFRD
może być niedoszacowane.
Pomimo że CFRD może się ujawnić w każdym
wieku, w tym również u niemowląt, jej częstość
zwiększa się z wiekiem. Według danych European
Epidemiologic Registry of Cystic Fibrosis
(ERCF) częstość występowania CFRD wynosi 5% w wieku 10–14 lat oraz 13% w wieku 15–19 lat.10 W badaniu z prospektywnym zbieraniem danych
przeprowadzonym w Irlandii częstość była podobna:
NGT stwierdzono u 69% badanych, IGT u 14% i CFRD u 17% badanych w wieku 10–19 lat.11 W badaniu duńskim u 50% chorych rozwinęła
się CFRD przed ukończeniem 30 lat.12 Według
danych jednego z amerykańskich ośrodków cukrzycę
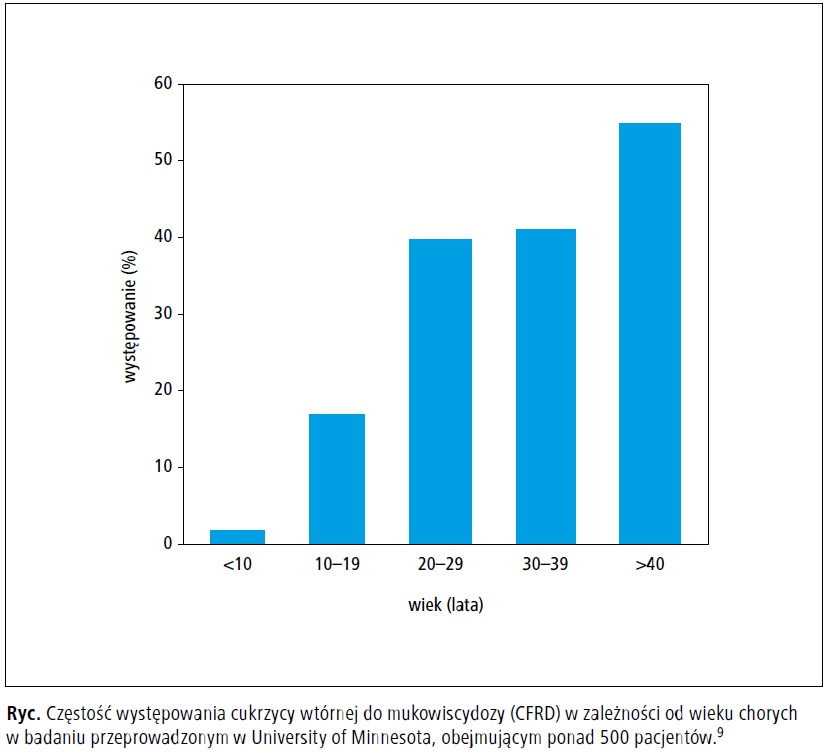
stwierdzano u <5% dzieci <10. roku życia,
15–20% nastolatków, około 40% chorych między
20. a 30. rokiem życia, a także u >50% chorych
>40. roku życia (ryc.).9
Patofizjologia CFRD
CFRD jest chorobą o złożonej patofizjologii. Pierwotny defekt – niedobór insuliny – obecny u prawie wszystkich pacjentów, wynika z jednoczesnego zniszczenia wysp trzustkowych i tkanki zewnątrzwydzielniczej. Jednak nie wszyscy chorzy na mukowiscydozę zachorują na cukrzycę, a metabolizm zależy od wielu czynników, w tym nasilenia zakażeń i stanu zapalnego, predyspozycji genetycznych, niedożywienia i prawdopodobnie rodzaju defektu kanału chlorkowego.
Zaburzenia czynności trzustki
Nieprawidłowe działanie kanałów chlorkowych w mukowiscydozie jest przyczyną produkcji lepspowodowakiej wydzieliny powodującej zatykanie przewodów, prowadzące do upośledzenia zewnątrzwydzielniczej czynności trzustki, postępującego włóknienia i stłuszczenia gruczołu. Skutkuje to zakłóceniem i zniszczeniem architektury wysp, prowadzącym do utraty wydzielniczej funkcji komórek ß i α oraz komórek produkujących polipeptyd trzustkowy. 13-15 Większość chorych na mukowiscydozę z cukrzycą lub bez cukrzycy traci około połowy komórek wysp trzustkowych. Niszczenie komórek ß w mukowiscydozie nie jest związane z autoimmunizacją, ponieważ autoprzeciwciała oraz antygeny zgodności tkankowej (human leukocytic antigens – HLA) związane z cukrzycą typu 1 występują z częstością podobną do częstości w populacji ogólnej.16,17 Mimo to, u nielicznych pacjentów rozpoznaje się zarówno cukrzycę typu 1, jak i mukowiscydozę.
Znaczenie niedoboru insuliny
Pierwotnym zaburzeniem w CFRD jest ciężki, ale nie całkowity niedobór insuliny. Praktycznie wszyscy chorzy z niewydolnością zewnątrzwydzielniczą trzustki w przebiegu mukowiscydozy – niezależnie od tego, czy chorują na cukrzycę czy nie – wykazują objawy niewydolności komórek ß.8,18 Stężenia insuliny i peptydu C na czczo są prawidłowe, ale występuje opóźnienie i zmniejszenie szczytu wydzielania insuliny w trakcie standardowego OGTT.19 Ten efekt jest silniej wyrażony przy zwiększaniu się glikemii.20-22 Opóźnione wydzielanie insuliny w OGTT jest związane z utratą pierwszej fazy wydzielania insuliny, która występuje nawet u tych chorych na mukowiscydozę, u których tolerancja glukozy jest prawidłowa.23 Upośledzone jest także wydzielanie glukagonu z uwagi na zniszczenie całych wysp trzustkowych.19-23
Znaczenie insulinooporności
U chorych na mukowiscydozę bez cukrzycy
wrażliwość na insulinę jest z reguły stała, chociaż
istnieją doniesienia o oporności na insulinę
zależnej od stopnia ciężkości choroby.24-28 Podczas
gdy u większości tych chorych w stabilnym stanie
zdrowia wrażliwość na insulinę jest zachowana,
zakażenia nasilają insulinooporność. U chorych na mukowiscydozę i cukrzycę występuje natomiast
niewielka insulinooporność spowodowana
zarówno zmniejszonym wychwytem glukozy przez
tkanki obwodowe, jak i słabym hamowaniem przez
insulinę wątrobowej produkcji glukozy.26,27
Insulinooporność nie odgrywa tak ważnej roli w patogenezie CFRD jak niedobór insuliny, mimo
to wydaje się, że ma dominujący wpływ w sytuacjach
stresowych, takich jak zaostrzenie choroby
płuc podczas zakażeń o ostrym przebiegu.
Genetyka CFRD
Mukowiscydoza jest spowodowana mutacją genu
regulatora przezbłonowego przewodnictwa (cystic
fibrosis transmembrane conductance regulator
– CFTR), kompleksu białek tworzących kanał
chlorkowy. Cukrzyca występuje głównie u osób z mutacjami powodującymi mukowiscydozę o ciężkim
przebiegu, z niewydolnością zewnątrzwydzielniczą
trzustki. CFTR występuje w komórkach ß,
gdzie jego rola nie została jeszcze poznana.29,30 W modelu zwierzęcym mukowiscydozy (fretka)
wykazano nieprawidłowe wydzielanie insuliny
od momentu narodzin, co może sugerować istotną
rolę CFTR w wydzielaniu insuliny.31 Spostrzeżenie
to potwierdzono również w małym badaniu
pilotażowym z udziałem chorych na mukowiscydozę, u których stwierdzono poprawę wydzielania
insuliny w odpowiedzi na doustną i dożylną postać
glukozy po podaniu nowego leku korygującego
funkcję CFTR.32
Częstsze występowanie cukrzycy typu 2 u monozygotycznych
bliźniąt, w porównaniu z bliźniętami
dizygotycznymi chorymi na mukowiscydozę,
33 oraz częstsze występowanie CFRD u chorych z dodatnim wywiadem w kierunku rodzinnego
występowania cukrzycy typu 2,34 a także związek z genami podatności na cukrzycę typu 234,35
sugeruje genetyczną zależność pomiędzy mukowiscydozą a cukrzycą typu 2. Opisano również
prawdopodobny związek pomiędzy CFRD a genami
związanymi z procesem stanu zapalnego, takimi
jak: czynnik martwicy nowotworów,36 białko
szoku termicznego37 oraz kalpaina 10.38 Na podstawie
tych obserwacji sformułowano hipotezę mówiącą o nadrzędnej roli częściowej utraty komórek
wysp trzustkowych w rozwoju CFRD, spowodowanej ich fizycznym zniszczeniem. Natomiast chorzy
ze stwierdzonym defektem wydzielania insuliny
lub zmniejszeniem wrażliwości na insulinę są
bardziej narażeni na rozwój cukrzycy, ponieważ
mają mniejszą zdolność kompensacji zredukowanej
liczby komórek ß.
| Tabela 3. Objawy kliniczne cukrzycy wtórnej do mukowiscydozy |
|---|
| niewyjaśniony wielomocz lub wzmożone pragnienie
brak przyrostu lub zmniejszenie masy ciała pomimo interwencji żywieniowych zbyt małe tempo wzrastania opóźnienie dojrzewania płciowego niewyjaśnione przewlekłe pogorszenie czynności płuc objawy mogą nie występować |
Objawy kliniczne CFRD
CFRD rozwija się skrycie. Objawy CFRD wymieniono w tabeli 3. Należy podkreślić, że większość chorych może nie wykazywać typowych objawów. Cukrzycowa kwasica ketonowa występuje rzadko, najprawdopodobniej z powodu zachowania wydzielania endogennej insuliny lub równoczesnego upośledzenia wydzielania glukagonu. Pierwsze objawy CFRD mogą wystąpić w sytuacjach nasilających insulinooporność, takich jak: ostre infekcyjne zapalenie płuc, leczenie GKS, przyjmowanie dużych ilości węglowodanów (ciągłe żywienie nocne). CFRD występuje często u osób po przeszczepieniu płuc, a ponieważ ta grupa chorych przed przeszczepieniem jest poważnie chora, w związku z tym rozwija się oporność na insulinę, a po przeszczepieniu pacjenci otrzymują leki diabetogenne, takie jak GKS lub inhibitory kalcyneuryny.39-42 CFRD występuje częściej u osób z chorobą wątroby w przebiegu mukowiscydozy.43
Przeżywalność i rokowanie
Zwiększona umieralność u chorych z CFRD
Od początku lat 80. w Stanach Zjednoczonych i w Europie udokumentowano większe ryzyko
zgonu u chorych na mukowiscydozą, u których
dodatkowo rozpoznano cukrzycę. Szczególnie na ryzyko wczesnego zgonu narażone były kobiety z CFRD.44-48 Przyczyną zgonu pacjentów z CFRD,
podobnie jak większości chorych na mukowiscydozę,
jest raczej niewydolność oddechowa niż powikłania
naczyniowe o charakterze makro- i mikroangiopatii,
które są przyczyną zgonu u chorych
na cukrzycę typu 1 i 2. Cukrzyca ma bezpośredni
wpływ na rozwój upośledzenia czynności płuc w przebiegu mukowiscydozy poprzez kataboliczny
efekt niedoboru insuliny, oddziaływania na stan
odżywienia oraz masę mięśniową,49-52 a także
negatywne działanie przewlekłej hiperglikemii
na czynność płuc,53-56 która w pewnym zakresie
sprzyja procesom zapalnym i ułatwia rozwój bakteriom.
W 2009 roku przedstawiono trendy umieralności w populacji chorych z CFRD na podstawie
badania dużej grupy chorych na mukowiscydozę,
obserwowanych długoterminowo w jednym
ośrodku od wczesnych lat 90.9 Między 1992 a 2008 rokiem wystąpiło znaczne i utrzymujące
się zmniejszenie ryzyka zgonów związanych z CFRD. We wczesnych latach 90. umieralność
wśród chorych z CFRD była 13,4 razy większa niż
wśród chorych bez cukrzycy. W 2008 roku wartość
ta zmalała do 3,5-krotnej różnicy, istotnej
jedynie u osób >30. roku życia. Nie obserwowano
już różnic w umieralności między płciami. Istotną
poprawę wskaźników umieralności związanej z CFRD uzyskano dzięki corocznym badaniom
przesiewowym w kierunku cukrzycy oraz wczesnemu
rozpoczynaniu leczenia insuliną.
Powikłania mikro- i makronaczyniowe
U chorych z CFRD opisywano powikłania mikronaczyniowe, które zwykle mają łagodny przebieg (istnieją jednak doniesienia o cięższych postaciach). W badaniu przeprowadzonym w Danii retinopatię stwierdzono u 36% osób chorujących na cukrzycę od ponad 10 lat.57 W innym badaniu przeprowadzonym w Stanach Zjednoczonych z udziałem 285 chorych z CFRD powikłania cukrzycy rzadko występowały przed upływem 10 lat od jej rozpoznania, natomiast po 10 latach trwania choroby u osób z CFRD z hiperglikemią na czczo mikroalbuminurię wykazano u 14% chorych, retinopatię u 16%, neuropatię u 55%, a gastropatię u 50% chorych.58 Nie wykazano natomiast powikłań mikronaczyniowych u chorych z CFRD, u których nigdy nie wystąpiła hiperglikemia na czczo.1 Dotychczas nie opisano zgonów chorych na mukowiscydozę z powodów powikłań makronaczyniowych. Jest to o tyle ważne, że ryzyko występowania takich powikłań determinuje leczenie i postępowanie u chorych na cukrzycę typu 1 i 2. Wiele z opracowanych wytycznych nie znajduje zastosowania w przypadku mukowiscydozy, a nawet mogą być szkodliwe. Stężenie cholesterolu w przebiegu mukowiscydozy jest z reguły zmniejszone, natomiast dość często stwierdza się izolowane zwiększenie stężenia trójglicerydów we krwi.59-63 Zwiększenie stężenia lipidów we krwi częściej występuje u chorych po przeszczepieniu płuc oraz u starszych pacjentów z mukowiscydozą z mutacjami powodującymi chorobę o mniej ciężkim przebiegu. Znaczenie kliniczne nieprawidłowego profilu lipidowego nie jest znane, choć wydaje się, że ma niekorzystny wpływ na przebieg choroby u starszych pacjentów.
Hipoglikemia
Hipoglikemia dość często występuje u chorych na mukowiscydozę z CFDR lub bez cukrzycy. W badaniu włoskim hipoglikemię na czczo stwierdzono u 14% spośród 129 dzieci i dorosłych chorych na mukowiscydozę, co wiązano z gorszym stanem klinicznym chorego (znaczne upośledzenie czynności płuc, częstsze hospitalizacje).28 W tej samej grupie chorych reaktywną hipoglikemię stwierdzono u 15% poddanych OGTT, podczas gdy w badaniu niemieckim u 6,3%.64 Prawdopodobnie jest to związane z opóźnieniem wydzielania insuliny. Mimo że w przebiegu mukowiscydozy produkcja glukagonu jest zmniejszona, u chorych stwierdza się prawidłową normalizację po hipoglikemii indukowanej insuliną, głównie z uwagi na utrzymaną produkcję katecholamin.32 Podobnie jak u wszystkich pacjentów leczonych insuliną, hipoglikemia jest groźnym stanem, a chorzy i ich rodziny powinni wiedzieć, jak przewidzieć jej wystąpienie, jak jej zapobiegać i jak ją leczyć.
Zwiększone ryzyko powikłań w stanie przedcukrzycowym
W kilku badaniach wykazano utajone pogarszanie się stanu klinicznego rok przed rozpoznaniem CFRD, w stanie przedcukrzycowym charakteryzującym się niedostateczną produkcją insuliny. 44,65-68 W badaniu prospektywnym wykazano, że pogorszenie czynności płuc w okresie 4 lat było najmniejsze u chorych z prawidłową tolerancją glukozy, większe u chorych z nieprawidłową tolerancją, a największe u osób chorych na mukowiscydozę i nieleczoną wczesną (bez stwierdzanej na czczo hiperglikemii) cukrzycę.66 Wyniki tego, a także innych badań sugerują, że pogorszenie czynności płuc zależy od stopnia niedoboru insuliny. 28 Zależność między katabolizmem białek, niedożywieniem a zgonem z powodu mukowiscydozy oraz silnym anabolicznym działaniem insuliny sprawia, że wpływ niedoboru insuliny na stan odżywienia i metabolizm może mieć w przypadku mukowiscydozy większe znaczenie niż metaboliczne skutki hiperglikemii. Dlatego chorzy mogą przez długi okres nie wykazywać żadnych objawów cukrzycy. Kataboliczne działanie niedoboru insuliny może mieć szczególne znaczenie u dzieci w okresie wzrastania.69-71
Badania przesiewowe w kierunku CFRD
Ważne znaczenie ma wykrywanie CFRD u chorych przed wystąpieniem jej objawów, ponieważ początek choroby często jest skryty. Obecnie jedynym badaniem przesiewowym uznawanym w rozpoznaniu CFRD jest standardowy test OGTT. Test należy wykonać co najmniej 8 h po ostatnim posiłku, po doustnym obciążeniu glukozą w dawce 1,75 g/kg mc. (maks. 75 g), a stężenie glukozy oznacza się po 2 h.
Doustny test obciążenia glukozą
Zgodnie z wytycznymi North American CFRD Guidelines
Committee badaniem przesiewowym z wyboru
do rozpoznawania CFRD jest OGTT.1 W porównaniu z OGTT, inne metody badań u chorych
na mukowiscydozę charakteryzuje mała wiarygodność, ponadto OGTT dostarcza długoterminowych
istotnych prognostycznie danych o możliwym przebiegu
cukrzycy, pozwala również na wczesne rozpoznanie
cukrzycy przy utrzymującym się jeszcze
prawidłowym stężeniu glukozy na czczo. U blisko 2/3
pacjentów z CFRD nie stwierdza się hiperglikemii
na czczo,9 dlatego zwiększoną glikemię można wykryć
jedynie w OGTT. Istotne jest wczesne rozpoznanie
hiperglikemii u chorych na mukowiscydozę,
ponieważ ta grupa jest bardziej narażona na wystąpienie
znacznego upośledzenia czynności płuc oraz
progresji w kierunku hiperglikemii na czczo,8 poza
tym szybkie rozpoczęcie leczenia insuliną poprawia
stan odżywienia tych chorych.72 OGTT pozwala
również na identyfikację chorych z upośledzoną
tolerancją glukozy. W dużym badaniu z udziałem
ponad 1000 chorych na mukowiscydozę z Niemiec i Austrii stwierdzono, że IFG, IGT oraz nieokreślona
hiperglikemia (INDET) są istotnymi czynnikami
rokowniczymi w CFRD.73
Cukrzyca w okresie ciąży stanowi zagrożenie
dla matki i płodu. U kobiet chorych na mukowiscydozę
cukrzyca ciężarnych rozwija się w dość
wczesnym okresie ciąży.6,7,74 OGTT należy wykonać
przed ciążą albo zaraz na początku ciąży, konieczne
jest również jego powtórzenie pod koniec
II i III trymestru.1
Wykazano również, że stężenie glukozy oznaczane w OGTT w innych punktach czasowych
może mieć większe prognostyczne znaczenie przy
określeniu klinicznego przebiegu CFRD niż stężenie
glukozy 2 godziny po obciążeniu. Należy zatem
rozważyć pomiary stężenia glukozy co pół godziny w czasie 2-godzinnego testu OGTT.55,73,75,76 Zaleca
się, aby pierwszy OGTT przeprowadzić przed
ukończeniem 10. roku życia. Mimo że cukrzyca w populacji ogólnej rzadko występuje u dzieci
<10. roku życia, to u 42–78% dzieci w wieku 9 lat
chorych na mukowiscydozę stwierdza się nieprawidłową
tolerancję glukozy.7,77,78 W badaniu prospektywnym z długofalową obserwacją chorych
przeprowadzonym w jednym z ośrodków North
American CF Center stwierdzono, że u dzieci w wieku 6–9 lat chorych na mukowiscydozę IGT
oraz nieokreślona hiperglikemia są niekorzystnymi
czynnikami rokowniczymi związanymi z dużym
ryzykiem rozwoju cukrzycy we wczesnym
wieku młodzieńczym.78 Z tego powodu w niektórych ośrodkach badania przesiewowe w kierunku
CFRD przeprowadza się już od 6. roku życia.
Oznaczenie HbA1c jako narzędzie diagnostyczne
W kilku badaniach wykazano, że oznaczenie HbA1c nie jest wiarygodne w diagnostyce CFRD z powodu wyników fałszywie ujemnych.8,44,79 Prawdopodobnie ma to związek ze zwiększonym obrotem erytrocytów spowodowanym stanem zapalnym. W jednym z badań zwiększone wartości HbA1c w chwili ustalenia rozpoznania cukrzycy stwierdzono tylko u 16% pacjentów z CFRD.8 Zwiększona wartość HbA1c potwierdza rozpoznanie hiperglikemii, natomiast prawidłowy wynik nie wyklucza zaburzeń metabolizmu glukozy.
Glikemia przygodna i glikemia na czczo oraz SMBG w diagnostyce CFRD
Mimo że hiperglikemia jest kryterium diagnostycznym cukrzycy, prawidłowe stężenie glukozy na czczo lub przygodne nie wyklucza rozpoznania cukrzycy u chorego na mukowiscydozę. W sytuacjach zwiększonego ryzyka, takich jak leczenie antybiotykami podawanymi dożylnie w warunkach domowych lub GKS, nocne żywienie przez gastrostomię, praktycznym podejściem jest wyjściowa samokontrola glikemii przez chorego za pomocą glukometru (SMBG). SMBG nie jest wystarczająco dokładnym badaniem, na podstawie którego można by rozpoznać cukrzycę, konieczne jest jej potwierdzenie w zalecanych badaniach przesiewowych wykonanych w labolatorium.
Ciągłe monitorowanie glikemii
Potwierdzono przydatność CGM u dzieci i młodzieży z CFRD w bezpiecznym i skutecznym prowadzeniu
leczenia insuliną.2 Znaczenie kliniczne
CGM u chorych na mukowiscydozę bez cukrzycy
ciągle pozostaje przedmiotem badań. CGM nie jest
natomiast wystarczająco dokładnym badaniem,
aby można je było wykorzystać w ustaleniu rozpoznania
cukrzycy.
Wiadomo, że u chorych na mukowiscydozę po posiłku występuje hiperglikemia, którą można wykryć za pomocą CGM jeszcze zanim wyniki
OGTT zmienią się z NGT na IGT lub cukrzycę,
jednak znaczenie kliniczne tych krótkotrwałych
epizodów hiperglikemii nie jest znane.75,80
Leczenie CFRD
Leczenie żywieniowe
Zalecenia żywieniowe dla chorych na mukowiscydozę z CFRD są znacząco inne niż dla chorych na cukrzycę typu 1 lub 2 (tab. 4.), co wynika z odmiennych potrzeb tej grupy pacjentów oraz mniejszego ryzyka wystąpienia chorób układu sercowo-naczyniowego u chorych na mukowiscydozę. Wszyscy chorzy na mukowiscydozę, w tym z CFRD, wymagają diety wysokokalorycznej, bogatej w sód i tłuszcze. Z tego powodu u większości pacjentów – ewentualnie z wyjątkiem starszych chorych na mukowiscydozę o umiarkowanym przebiegu, z nadwagą – ograniczenie podaży kalorii zwykle jest przeciwwskazane. Dla pacjentów leczonych insuliną ważne jest obliczanie ilości węglowodanów w celu ustalenia należnej dawki insuliny. Pokrycie należnego zapotrzebowania na insulinę przy spożyciu większej ilości gazowanych słodzonych napojów może być trudne, dlatego nie zaleca się ich spożywania.
Leczenie insuliną
Niedobór insuliny jest istotą etiopatogenezy CFRD, dlatego insulina jest jedynym lekiem zalecanym w jej leczeniu.1 Insulinoterapia może pomóc w stabilizacji czynności płuc i poprawić stan odżywienia chorych na CFRD.9,72,81 Główne założenia insulinoterapii w CFRD opisano w tabeli 5. U chorych na mukowiscydozę w stabilnym stanie zdrowia, bez dodatkowych obciążeń, podstawowe zapotrzebowanie na insulinę jest umiarkowane z uwagi na wciąż obecną endogenną produkcję własnej insuliny oraz prawdopodobnie zmniejszenie stężenia glukagonu (u młodzieży i dorosłych przeciętna dawka insuliny wynosi <0,5–0,8 j./ kg mc./24 h).82,83 U chorych z hiperglikemią na czczo zaleca się intensywną insulinoterapię z wykorzystaniem osobistych pomp insulinowych lub połączenia długo działającego analogu insuliny jako insuliny podstawowej (bazowej) i szybko działającego analogu insuliny (bolus) w dawkach należnych na pokrycie spożywanych węglowodanów oraz kontroli epizodów hiperglikemii. W przypadku chorych z CFRD bez hiperglikemii na czczo obowiązujący standard leczenia obejmuje wstrzyknięcia przed posiłkiem szybko działającego analogu insuliny mające zabezpieczyć chorych przed przewlekłą utratą masy ciała.72 Z uwagi na zależność między stanem odżywienia a ogólnym przeżyciem chorych na mukowiscydozę, nadrzędną korzyścią insulinoterapii może być działanie anaboliczne insuliny. Dlatego celem leczenia jest podawanie chorym największych bezpiecznych, tolerowanych przez nich dawek.
| Tabela 4. Zalecenia żywieniowe dla chorych na cukrzycę typu 1,typu 2 i cukrzycę wtórną do mukowiscydozy | ||
|---|---|---|
| Cukrzyca typu 1 i 2 | CFRD | |
| zapotrzebowanie energetyczne (kcal) | ≤100% normalnego zapotrzebowania dla wieku i płci– często konieczne przestrzeganie ograniczeń kalorycznych, aby zapobiegać rozwojowi nadwagi | zwykle wymagane pokrycie120–150% (lub więcej) normalnego zapotrzebowania dla wieku i płci, aby zapobiegać niedoborowi masy ciała |
| tłuszcze | <30–35% całkowitego zapotrzebowania energetycznego | 40% całkowitego zapotrzebowania energetycznego |
| węglowodany | 45–60% całkowitego zapotrzebowania energetycznego | 40–50% całkowitego zapotrzebowania energetycznego |
| błonnik | ok. 3,3 g błonnika na megadżul (MJ) | zalecany u prawidłowo odżywionych, ale u źle odżywionych może zmniejszać ilość spożytych składników energetycznych |
| białko | 15–20% całkowitego zapotrzebowania energetycznego,ok. 1–2 g/kg mc./24 h | 200% zalecanego spożycia |
| sól | zakres 1000–1500 mg/24 h | zwiększone zapotrzebowanie – nieograniczona podaż |
| CFRD – cukrzyca wtórna do mukowiscydozy | ||
Doustne leki przeciwcukrzycowe
Obecnie nie zaleca się doustnych leków przeciwcukrzycowych w leczeniu CFRD. W przeglądzie Cochrane nie znaleziono badań z randomizacją dotyczących tego zagadnienia, poza badaniem CFRDT, w którym wykazano, że pobudzający wydzielanie insuliny repaglinid nie był skuteczny w utrzymaniu trwałego zwiększenia masy ciała u chorych na CFRD bez hiperglikemii na czczo.72 Leki zmniejszające insulinooporność raczej nie są skuteczne w CFRD, ponieważ insulinooporność nie jest głównym czynnikiem etiologicznym. Działania niepożądane aktualnie dostępnych leków uwrażliwiających na działanie insuliny, takie jak upośledzenie czynności przewodu pokarmowego (metformina) lub osteoporoza (pochodne tiazolidynedionu), są nie do zaakceptowania u chorych na mukowiscydozę. Obecnie brak wiarygodnych wyników badań potwierdzających skuteczność leków pobudzających układ inkretynowy (inkretyn), takich jak agonista peptydu glukagonopodobnego 1 (GLP-1) oraz inhibitorów peptydazy dipeptydylowej 4 (DPP-4), u chorych na mukowiscydozę. Nie wydaje się również, żeby pacjenci z CFRD mogli skorzystać na leczeniu tymi lekami, ponieważ mechanizm ich działania polega na opóźnianiu opróżniania żołądka i zmniejszaniu stężenia glukagonu.
| Tabela 5. Zasady leczenia insuliną w cukrzycy wtórnej do mukowiscydozy (CFRD) | |
|---|---|
| główne założenia | Chorzy na CFRD bez chorób towarzyszących wymagają stosowaniadawki 0,5–0,8 j./kg mc./24 h. W czasie chorób towarzyszących o ostrymprzebiegu dawkę należy zwiększyć. Z uwagi na kataboliczny skutek niedoboru insuliny, zaleca siępodawanie chorym jak największych, bezpiecznych dawek insuliny. Należy zastosować optymalny schemat insulinoterapii dopasowanydo stylu życia chorego oraz leczenia mukowiscydozy. |
| insulina podstawowa | Insulina podstawowa – na ogół dawką docelową jest ok. 0,25 j./kg mc./24 h; leczenie należy rozpocząć od podania połowy tej dawki,a następnie zwiększać ją na podstawie stężenia glukozy na czczo. |
| insulina posiłkowa | Zwykle dawka początkowa wynosi 0,5–1 j. szybko działającego analogu insuliny na każde 15 g spożywanych węglowodanów. Mogą być potrzebne wstrzykiwacze (peny) lub strzykawki umożliwiające dawkowanie co pół jednostki. Dostosowywanie dawki poprzez zwiększanie o 0,5 j. na 15 g węglowodanów, aż do osiągnięcia docelowych wartości glikemii 2 h po posiłku. U chorych w bardzo młodym wieku lub chorych, którzy nie są pewni,ile zjedzą z powodu nudności lub gastroparezy, insulinę można podać bezpośrednio po posiłku (chociaż zawsze lepszym postępowaniem jest podanie przed posiłkiem, jeśli to możliwe). U pacjentów z CFRD bez hiperglikemii na czczo insulinoterapię można prowadzić według dwóch schematów: jedynie insulina posiłkowa wstrzykiwana bezpośrednio przed posiłkami lub tylko wstrzyknięcie insuliny podstawowej, w zależności od indywidualnych cech pacjenta(np. zwyczajów żywieniowych). |
| dawki korekcyjne (wrażliwość) | Początkowa dawka przed posiłkiem zwykle wynosi 0,5–1 j. szybko działającego analogu insuliny na każde 50 mg/dl (2,8 mmol/l) glukozy powyżej 150 mg/dl (8,3 mmol/l), którą następnie należy dostosowywać zgodnie z potrzebami. |
| zapotrzebowanie związane z ciągłym żywieniem nocnym | Zapotrzebowanie związane z ciągłym żywieniem nocnym często pokrywa jedna dawka insuliny krótko działającej z dodatkiem insuliny izofanowej (np. Humulin N, Protaphane, Novolin N, Insulatard,Isophane itp). Insulina krótko działająca zabezpiecza pierwsze 4 h(pierwszą połowę), a insulina izofanowa (NPH) kolejne 4 (drugą połowę nocnego żywienia). Dawka początkowa: należy obliczyć całkowitą ilość węglowodanów w pożywieniu, ustalić całkowitą dawkę insuliny, stosując przelicznik liczby potrzebnych jednostek insuliny do ilości węglowodanów (zwykle0,5–1 j. insuliny na każde 15 g spożywanych węglowodanów) i połowę należnej dawki podać w postaci insuliny krótko działającej, a drugą w postaci insuliny izofanowej (NPH). Pomiar glikemii 4 h po rozpoczęciu żywienia służy dobraniu dawki insuliny krótko działającej, natomiast pomiar po jego zakończeniu dawki insuliny izofanowej. Niekiedy konieczne jest również podanie na początku małej dawki szybko działającego analogu insuliny. Powinno się traktować ciągłe żywienie nocne jako „długi posiłek”,dlatego nie należy zmieniać dawki insuliny podstawowej, a pacjent powinien otrzymywać dodatkowo odpowiednią insulinę tylko w trakcie żywienia. |
| leczenie w warunkach ograniczonych zasobów | Jeżeli analogi insuliny nie są dostępne, w leczeniu CFRD można wykorzystać insulinę izofanową (np. Humulin N, Protaphane, Novolin N,Insulatard, Isophane, itd) i krótko działającą, ale należy się starać unikać późnej poposiłkowej hipoglikemii. Możliwy schemat insulinoterapii u chorych spożywających 3 posiłki i 3 przekąski w ciągu dnia zakłada podawanie insuliny izofanowej (NPH) przed snem, a krótko działającej przed śniadaniem, obiadem i kolacją. |
Leczenie szpitalne chorych na CFRD
Podczas choroby towarzyszącej o ostrym przebiegu u chorych na mukowiscydozę występuje zwiększone ryzyko hiperglikemii.85,86 Podczas gdy dane dotyczące innych populacji sugerują, że intensywna insulinoterapia może być korzystna u pacjentów hospitalizowanych, w żadnym badaniu nie oceniano korzyści wynikających z utrzymania normoglikemii u hospitalizowanych chorych na mukowiscydozę. U osób z wcześniej rozpoznaną cukrzycą zapotrzebowanie na insulinę podczas ostrej choroby towarzyszącej może być znacznie większe, niektórzy chorzy wymagają nawet 4-krotnego zwiększenia wyjściowej dawki. Dawka insuliny musi być szybko zmniejszana w okresie zdrowienia, aby uniknąć hipoglikemii, chociaż okres ten może trwać kilka miesięcy.85 U chorych z prawidłowym stężeniem glukozy przed ostrą chorobą, glikemia może powrócić do wartości prawidłowych po ustąpieniu ostrej choroby, istnieje jednak prawdopodobieństwo nawrotu hiperglikemii z każdą kolejną chorobą o ostrym przebiegu.
Leczenie CFRD z upośledzoną tolerancją glukozy
Wyniki badań bez grupy kontrolnej, obejmujących małą liczbę chorych wykazały, że leczenie insuliną może być korzystne dla chorych z IGT.81,87-89 Obecnie nie ma jednak wystarczających danych świadczących o korzyściach insulinoterapii u chorych na mukowiscydozę bez rozpoznanej cukrzycy. Zagadnienie to pozostaje nadrzędnym celem wielu aktualnie prowadzonych badań.1
Zalecenia
ISPAD oficjalnie poparło wytyczne z 2010 roku sponsorowane przez ADA i Cystic Fibrosis Foundation, zaakceptowane także przez Pediatric Endocrine Society, przedstawione jako American Diabetes Association Position Statement.1
Kryteria diagnostyczne CFRD
• CFRD należy rozpoznać, gdy chory na mukowiscydozę
po raz pierwszy spełnił kryteria diagnostyczne
cukrzycy, nawet jeśli w kolejnych badaniach
obserwuje się stopniowe zmniejszanie
hiperglikemii (E).
• W czasie stabilnej fazy choroby (bez ostrej choroby
towarzyszącej) rozpoznanie CFRD ustala
się na podstawie standardowych kryteriów
ADA (E):
– glikemia na czczo ≥126 mg/dl (≥7,0 mmol/l) lub
– glikemia 2 h po obciążeniu w trakcie OGTT
≥200 mg/dl (≥11,1 mmol/l), lub
– HbA1c ≥6,5% (48 mmol/l [prawidłowa wartość
HbA1c nie wyklucza rozpoznania CFRD]), lub
– przygodna glikemia ≥200 mg/dl (11,1 mmol/l)
oraz objawy cukrzycy.
• Rozpoznanie CFRD można ustalić w czasie
występowania towarzyszącej choroby o ostrym
przebiegu (dożylna antybiotykoterapia w szpitalu
lub w domu, podawanie GKS ogólnie), jeśli
glikemia na czczo
126 mg/dl (
7,0 mmol/l) lub 2 h po posiłku
200 mg/dl (11,1 mmol) utrzymuje
się przez ponad 48 h (E).
• Do rozpoznania CFRD u chorych żywionych dojelitowo w sposób ciągły konieczne jest stwierdzenie w 2 różnych dniach glikemii w czasie
wlewu i po jego zakończeniu
200 mg/dl
(11,1 mmol) (E).
• Cukrzycę ciążową rozpoznaje się na podstawie
wytycznych International Association of Diabetes
and Pregnancy Study Group,90 zgodnie z którymi cukrzycę stwierdza się, gdy glikemia w godzinie 0, 1 i 2 podczas OGTT z użyciem
75 g glukozy wynosi:
– na czczo ≥92 mg/dl (≥5,1 mmol/l) lub
– 1 h po obciążeniu ≥180 mg/dl (≥10 mmol/l), lub
– 2 h po obciążeniu ≥153 mg/dl (≥8,5 mmol/l).
• Rozpoznanie cukrzycy ciążowej nie jest jednoznaczne z rozpoznaniem CFRD, ale wymaga
przeprowadzenia badań przesiewowych
6–12 tygodni po zakończeniu ciąży (E).
• Nie jest konieczne rozróżnienie CFRD z hiperglikemią
na czczo od CFRD bez hiperglikemii
na czczo (B).
Badania przesiewowe
• Nie zaleca się oznaczania HbA1c do wykrywania
CFRD (B).
• Badaniem przesiewowym w rozpoznawaniu
CFRD jest OGTT z oceną glikemii 2 h po obciążeniu
glukozą w dawce 1,75 g/kg mc. (maks.
75 g [E]).
• Zaleca się rozpoczęcie przeprowadzania corocznych
badań w kierunku CFRD u wszystkich
chorych na mukowiscydozę, jeszcze przed ukończeniem
10. roku życia (B).
• U chorych na mukowiscydozę z zaostrzeniem
choroby układu oddechowego, którzy wymagają
dożylnej antybiotykoterapii i/lub podawania
GKS ogólnoustrojowo, należy wykonać badania
przesiewowe w kierunku CFRD z oznaczaniem
glikemii na czczo oraz 2 h po posiłku przez
pierwsze 48 h zaostrzenia (E).
• U chorych na mukowiscydozę żywionych dojelitowo
zaleca się oznaczenie stężenia glukozy w trakcie oraz bezpośrednio po zakończeniu
wlewu enteralnego, na początku karmienia
przez gastrostomię oraz raz w miesiącu podczas
żywienia w domu. Zwiększone stężenie
glukozy stwierdzane w ramach SMBG wymaga
potwierdzenia w formalnym badaniu glikemii
przeprowadzonym w certyfikowanym laboratorium
(E).
•Należy przeprowadzić badania w kierunku
CFRD z oznaczeniem glikemii 2 h po obciążeniu
75 g glukozy w teście OGTT u kobiet chorych
na mukowiscydozę planujących ciążę lub
będących w ciąży, o ile nie wykonywano u nich
badań w kierunku CFRD w ciągu ostatnich 6 miesięcy (E).
• Zaleca się wykonywanie badań przesiewowych w kierunku cukrzycy ciążowej między
12. a 16. oraz 24. a 28. tygodniem ciąży u kobiet
chorych na mukowiscydozę, u których
wcześniej nie stwierdzono CFRD, wykonując
OGTT z 75 g glukozy, z oznaczeniem glikemii w godzinie 0, 1 i 2 (E).
• Po okresie ciąży badanie przesiewowe w kierunku
CFRD u kobiet z cukrzycą ciążową (cukrzyca
rozpoznana po raz pierwszy w czasie ciąży) należy
wykonać między 6. a 12. tygodniem od jej
zakończenia, wykorzystując oznaczenie glukozy
po obciążeniu 75 g glukozy w 2-godzinnym teście
OGTT (E).
• Chorych na mukowiscydozę przygotowywanych
do przeszczepienia należy poddać badaniom
przesiewowym w kierunku CFRD z wykorzystaniem
testu OGTT, o ile nie wykonywano
tych badań w ostatnich 6 miesiącach. Stężenie
glukozy należy w tym wypadku monitorować w trakcie całego okresu pooperacyjnego do momentu
wypisania chorego ze szpitala. Wytyczne
dotyczące pacjentów, którzy w chwili wypisu
ze szpitala nie spełniają kryteriów rozpoznania
CFRD, są takie same jak dla pozostałych
chorych na mukowiscydozę (E).
Leczenie CFRD
• W optymalnych warunkach pacjentów z CFRD
powinien kontrolować raz na 3 miesiące wykwalifikowany,
wielospecjalistyczny zespół z doświadczeniem w leczeniu mukowiscydozy i cukrzycy (E).
• Pacjentów z CFRD należy objąć ciągłym programem
edukacyjnym w zakresie samokontroli
cukrzycy, spełniającym krajowe standardy (E).
• W leczeniu CFRD należy stosować insulinę (A).
• Doustne leki przeciwcukrzycowe nie są tak skuteczne
jak insulina w poprawie stanu odżywienia i metabolicznego chorych na CFRD, a ich
stosowanie należy ograniczyć jedynie do badań
naukowych (A).
• U chorych na CFRD zaleca się samodzielną
kontrolę glikemii przynajmniej 3 razy dziennie w czasie leczenia insuliną (E). Dla wielu
chorych pomiar 4–8 lub więcej razy dziennie
jest wystarczający, zależnie od pory posiłków,
ćwiczeń fizycznych, występowania powikłań
ze strony przewodu pokarmowego (takich jak
gastropareza) oraz od obecności chorób towarzyszących o ostrym przebiegu (E).
• U chorych na CFRD należy dążyć do osiągnięcia
stężeń glukozy określonych w wytycznych
ADA dla wszystkich chorych na cukrzycę, zakładając
potrzebę indywidualizacji terapii, bowiem u niektórych pacjentów wartości docelowe
mogą być mniejsze lub większe niż ustalone w wytycznych (E).
• W ramach monitorowania leczenia insuliną u chorych na CFRD zaleca się oznaczanie
HbA1c co 3 miesiące (E):
– u większości chorych na CFRD należy dążyć
do HbA1c <7% (53 mmol/mol), w celu zmniejszenia
ryzyka powikłań o charakterze mikronaczyniowym;
należy jednak uwzględnić potrzebę
indywidualizacji terapii, gdyż u niektórych
pacjentów wskazane są większe lub mniejsze
wartości (B).
• U chorych z CFRD zaleca się stosowanie wytycznych
żywieniowych jak dla wszystkich chorych
na mukowiscydozę, opracowanych przez
CF Foundation na podstawie wiarygodnych
danych (E).
• Chorym na CFRD zaleca się umiarkowany wysiłek
aerobowy co najmniej 150 min/tydzień (E).
Powikłania
• Ważna jest edukacja chorych na CFRD i ich
opiekunów w zakresie objawów, profilaktyki i leczenia hipoglikemii, w tym stosowania glukagonu
(E).
• U chorych na CFRD należy kontrolować ciśnienie
tętnicze w trakcie każdej rutynowej wizyty
lekarskiej zgodnie z zaleceniami ADA. U pacjentów, u których stwierdzono ciśnienie skurczowe
≥130 mm Hg lub ciśnienie rozkurczowe
≥80 mm Hg lub ciśnienie >90. centyla dla płci i wieku dla populacji pediatrycznej należy powtórzyć
pomiar ciśnienia tętniczego w innym
dniu w celu potwierdzenia rozpoznania nadciśnienia
tętniczego (E).
• Zaleca się, aby po upływie 5 lat od rozpoznania
CFRD przeprowadzać coroczne badania w kierunku mikroangiopatii cukrzycowej. Jeśli
dokładny czas rozpoznania CFRD jest nieznany,
badania te należy rozpocząć w momencie
stwierdzenia po raz pierwszy hiperglikemii
na czczo (E).
• Pacjentów z CFRD i nadciśnieniem tętniczym
lub powikłaniami naczyniowymi o charakterze
mikroangiopatii należy leczyć zgodnie z wytycznymi ADA dla wszystkich chorych
na cukrzycę, nie stosuje się jednak u nich ograniczeń w spożyciu sodu oraz, na ogół, również
białka (E).
• Coroczne oznaczanie profilu lipidowego zaleca
się chorym na CFRD z niewydolnością zewnątrzwydzielniczą
trzustki lub w przypadku
współistnienia takich czynników ryzyka, jak
otyłość, rodzinne występowanie choroby wieńcowej
lub leczenie immunosupresyjne po przeszczepieniu
(E).
Konflikt interesów: Autorzy nie zgłosili konfliktu interesów.
Piśmiennictwo:
1. Moran A., Brunzell C., Cohen R.C., et al.: Clinical care guidelines for CFRD: recommendations from the cystic fibrosis foundation, the American diabetes association and the pediatric endocrine society. Diabetes Care, 2010; 33: 2697–27082. O’Riordan S.M., Hindmarsh P., Hill N.R., et al.: Validation of continuous glucose monitoring in children and adolescents with cystic fibrosis: a prospective cohort study. Diabetes Care, 2009; 32: 1020–1022
3. Moreau F., Weiller M.A., Rosner V., et al.: Continuous glucose monitoring in CF patients according to the glucose tolerance. Horm. Metab. Res., 2008; 40: 502–506
4. Frohnert B., Ode K.L., Moran A., et al.: Impaired fasting glucose in cystic fibrosis. Diabetes Care, 2010; 33: 2660–2664
5. Scheuing N., Holl R.W., Dockter G., et al.: Diabetes in cystic fibrosis: Multicenter screening results based on current guidelines. PLoS One, 2013; 8: e81 545 doi:10.1371/journal. pone.0 081 545
6. Hardin D.S., Rice J., Cohen R.C., Ellis K.J., Nick J.A.: The metabolic effects of pregnancy in cystic fibrosis. Obstet. Gynecol., 2005; 106: 367–375
7. Martin-Frias M., Lamas Ferreiro A., Enes Romero P., Cano Gutierrez B., Barrio Castellanos R.: Abnormal glucose tolerance in prepubertal patients with cystic fibrosis. Ann. Pediatr. (Barc.), 2012; 77: 339–343
8. Lanng S., Hansen A., Thorsteinsson B., Nerup J., Koch C.: Glucose tolerance in cystic fibrosis: a five year prospective study. BMJ, 1995; 311: 655–659
9. Moran A., Dunitz J., Nathan B., Saeed A., Holme B., Thomas W.: Cystic fibrosis related diabetes: current trends in prevalence, incidence and mortality. Diabetes Care, 2009; 32: 1626–1631
10. Koch D., Rainisio M., Madessani U., et al.: Presence of cystic fibrosis-related diabetes mellitus is tightly Lidek to poor lung function in patients with cystic fibrosis: data from the European epidemiologic registry of cystic fibrosis. Pediatr. Pulmonol., 2001; 32: 343–350
11. O’Riordan S.M., Hoey H., Costigan C.: Demographics and prevalence of glucose intolerance and cystic fibrosis related diabetes in 167 cystic fibrosis children. Diabetes, 2006; 55: A224
12. Lanng S.: Glucose intolerance in cystic fibrosis patients. Paediatr. Respir. Rev., 1991; 2: 253–259
13. Iannucci A., Mukai K., Johnson D., Burke B.: Endocrine pancreas in cystic fibrosis: an immunohistochemical study. Hum. Pathol., 1984; 15: 278–284
14. Lohr M., Goertchem P., Nizze H., et al.: CF associated islet changes may provide a basis for diabetes. Virchows Arch. A. Pathol. Anat., 1989; 414: 179–185
15. Couce M., O’Brien T.D., Moran A., Roche P.C., Butler P.C.: Diabetes mellitus in CF is characterized by islet amyloidosis. J. Clin. Endocrinol. Metab., 1996; 81: 1267–1272
16. Gottlieb P.A., Yu L., Babu S., et al.: No relation between cystic-fibrosis related diabetes and type 1 diabetes autoimmunity. Diabetes Care, 2012; 35: e1
17. Bismuth E., Laborde K., Taupin P., et al.: Glucose tolerance and insulin secretion, morbidity and death in patients with CF. J. Pediatr., 2008; 152: 540–545
18. Holl R.W., Wolf A., Thon A., et al.: Insulin resistance with altered secretory kinetics and reduced proinsulin in CF patients. J. Pediatr., Gastroenterol. Nutr., 1997; 25: 188–193
19. Lanng S., Thorsteinsson B., Roder M.E., et al.: Pancreas and gut hormone responses to oral glucose and intravenous glucagon in cystic fibrosis patients with normal, impaired, and diabetic glucose tolerance. Acta Endocrinol., 1993; 128: 207–214
20. DeSchepper J., Dab I., Derde M.P., Loeb H.: Oral glucose tolerance testing in cystic fibrosis: correlations with clinical parameters and glycosylated hemoglobin determinations. Eur. J. Pediatr., 1991; 150: 403–406
21. Hamdi I., Payne S., Barton D., et al.: Genotype analysis in CF in relation to the occurrence of diabetes mellitus. Clin. Genet., 1993; 43: 186–189
22. Yung B., Noormohamed F.H., Kemp M., Hooper J., Lant A.F., Hodson M.E.: CFRD: the role of peripheral insulin resistance and beta-cell dysfunction. Diabet. Med., 2002; 19: 221–226
23. Moran A., Diem P., Klein D.J., Levitt M.D., Robertson R.P.: Pancreatic endocrine function in cystic fibrosis. J. Pediatr., 1991; 118: 715–723
24. Cucinotta D., De Luca F., Gigante A., et al.: No changes of insulin sensitivity in cystic fibrosis patients with different degrees of glucose tolerance: an epidemiological and longitudinal study. Eur. J. Endocrinol., 1994; 130: 253–258
25. Lanng S., Thorsteinsson B., Roder M.E., Nerup J., Koch C.: Insulin sensitivity and insulin clearance in CF patients with normal and diabetic glucose tolerance. Clin. Endocrinol., 1994; 41: 217–223
26. Hardin D.S., LeBlance A., Para L., Seilheimer D.K.: Hepatic insulin resistance and defects in substrate utilization in cystic fibrosis. Diabetes, 1999; 48: 1082–1087
27. Moran A., Pyzdrowski K., Weinreb M.D., et al.: Insulin sensitivity in cystic fibrosis. Diabetes, 1994; 43: 1020–1026
28. Battezatti A., Mari A., Zazzerson L., et al.: Identification of insulin secretory defects and insulin resistance during oral glucose tolerance test in a cohort of cystic fibrosis patients. Eur. J. Endocrinol., 2011; 165: 69–76
29. Edlund A., Huhn M., Flodstrom-Tullberg M., Eliasson L.: Active CFTR channels are important for insulin and glucagon secretion. Acta Physiologica, 2010; 198 (Suppl. 677P-MON-66)
30. Boom A., Lybaert P., Pollet J.F., et al.: Expression and localization of CFTR in the rat endocrine pancreas. Endocr. Pract., 2008; 32: 197–205
31. Olivier A.K., Yi Y., Sun X., et al.: Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J. Clin. Invest., 2012; 122: 3755–3768
32. Bellin M.D., Laguna T., Leschyshyn J., et al.: Proofof-concept pilot: insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR. Pediatr. Diabetes, 2013; 14: 417–421
33. Blackman S.M., Hsu S., Vanscoy L.L., et al.: Genetic modifiers play a substantial role in diabetes complicating cystic fibrosis. J. Clin. Endocrinol. Metab., 2009; 94: 1302–1309
34. Blackman S.M., Hsu S., Ritter S.E., et al.: A susceptibility gene for type 2 diabetes confers substantial risk for diabetes complicating cystic fibrosis. Diabetologia, 2009; 52: 1858–1865
35. Blackman S.M., Commander C.W., Watson C., et al.: Genetic modifiers of cystic fibrosis-related diabetes. Diabetes, 2013; 62: 3627–3635
36. Lanng S., Thorsteinsson B., Pociot F., et al.: Diabetes mellitus in cystic fibrosis: genetic and immunological markers. Acta Paediatr., 1993; 82: 150–154
37. Jensen P., Johansen H.K., Carmi P., Hoiby N., Cohen I.R.: Autoantibodies to pancreatic hsp60 precede the development of gkucose intolerance in patients with CF. J. Autoimmun., 2001; 17: 165–172
38. Derbel S., Doumaguet C., Hubert D., et al.: Calpain 10 and development of diabetes mellitus in cystic fibrosis. J. Cyst. Fibros., 2006; 5: 47–51
39. Braun A.T., Merlo C.A.: Cystic fibrosis and lung transplantation. Curr. Opin. Pulm. Med., 2011; 17: 467–472
40. Hadjiliadis D., Madill J., Chaparro C., et al.: Incidence and prevalence of diabetes mellitus in patients with cystic fibrosis undergoing lung transplantation before and after lung transplantation. Clin. Transplant., 2005; 19: 773–778
41. Bradbury R.A., Shirkhedkar D., Glanveill A.R., Campbell L.V.: Prior diabetes mellitus is associated with increased morbidity in CF patients undergoing bilateral lung transplantation: an „orphan” area?Aretrospective case-control study. Intern. Med. J., 2009; 39: 384–388
42. van Meerkerk G., van de Graff E.A., Kwakkelvan Erp J.M., et al.: Diabetes before and after lung transplantation in patients with cystic fibrosis and other lung diseases. Diabet. Med., 2012; 29: e159–e162
43. Marshall B.C., Butler S.M., Stoddard M., Moran A.M., Liou T.G., Morgan W.J.: Epidemiology of cystic fibrosis-related diabetes. J. Pediatr., 2005; 146: 681–687
44. Finkelstein S.M., Wielinski C.L., Elliott G.R., et al.: Diabetes mellitus associated with cystic fibrosis. J. Pediatr., 1988; 112: 373–377
45. Rosenecker J., Hofler R., Steinkamp G., et al.: Diabetes mellitus in patients with cystic fibrosis: the impact of diabetes mellitus on pulmonary function and clinical outcome. Eur. J. Med. Res., 2001; 6: 345–350
46. Milla C.E., Billings J., Moran A.: Diabetes is associated with dramatically decreased survival in female but not male subjects with cystic fibrosis. Diabetes Care, 2005; 28: 2141–2144
47. Sims E.J., Green M.W., Mehta A.: Decreased lung function in female but not male subjectswith established cystic fibrosis-related diabetes. Diabetes Care, 2005; 28: 1581–1587
48. Chamnan P., Shine B.S.F., Haworth C., Bilton D., Adler A.I.: Diabetes as a determinant of mortality in cystic fibrosis. Diabetes Care, 2010; 33: 311–316
49. Kien C.L., Zipf W.B., Horswill C.A., Denne S.C., McCoy K.S., O’Dorsio T.M.: Effects of feeding on protein turnover in healthy children and in children with cystic fibrosis. Am. J. Clin. Nutr., 1996; 64: 608–614
50. Hardin D.S., Leblanc A., Lukenbaugh S., Para L., Seilheimer D.K.: Proteolysis associated with insulin resistance in cystic fibrosis. Pediatrics, 1998; 101: 433–437
51. Moran A., Milla C., DuCret R., Nair K.S.: Protein metabolism in clinically stable adult CF patients with abnormal glucose tolerance. Diabetes, 2001; 50: 1336–1343
52. Moran A., Basu R., Milla C., Jensen M.: Insulin regulation of free fatty acid kinetics in adult cystic fibrosis patients with impaired glucose tolerance. Metabolism, 2004; 53: 1467–1472
53. Brennan A.L., Gyi K.M., Wood D.M., et al.: Airway glucose concentrations and effect on growth of respiratory pathogens in CF. J. Cyst. Fibros., 2007; 6: 101–109
54. Ntimbane T., Krishnamoorthy P., Huot C., et al.: Oxidative stress and cystic fibrosis-related diabetes: a pilot study in children. J. Cyst. Fibros., 2008; 7: 373–384
55. Suratwala D., Chan J.S.H., Kelly A., et al.: Nocturnal hypoxemia and glucose tolerance in children with cystic fibrosis. Thorax, 2011; 66: 574–578
56. Hunt W.R., Zughaier S.M., Guentert D.E., et al.: Hyperglycemia impedes lung bacterial clearance in a murine model of cystic-fibrosis-related diabetes. Am. J. Physiol. Lung Cell Mol. Physiol. 2014; 306: L43–L49
57. Andersen H.U., Lanng S., Tressler T., Laugesen C.S., Mathiesen E.R.: Cystic fibrosis-related diabetes: the presence of microvascular diabetes complications. Diabetes Care, 2006; 29: 2660–2663
58. Schwarzenberg S.J., Thomas W., Olsen T.W., et al.: Microvascular complications in cystic fibrosis-related diabetes. Diabetes Care, 2007; 30: 1056–1061
59. Figueroa V., Milla C., Parks E.J., Schwarzenberg S.J., Moran A.: Abnormal lipid levels in cystic fibrosis. Am. J. Clin. Nutr., 2002; 75: 1005–1011
60. Georgiopolou W., Denker A., Bishop K.L., et al.: Metabolic abnormalities in adults with CF. Respirology, 2010; 15: 823–829
61. Ishimo M.C., Belson L., Ziai S., et al.: Hypertriglyceridemia is associated with insulin levels in adult cystic fibrosis patients. J. Cyst. Fibros., 2013; 12: 271–276
62. Nash E.F., Stephenson A., Helm E.J., et al.: Impact of lung transplantation on serum lipids in adults with cystic fibrosis. J. Heart Lung Transplant., 2011; 30: 188–193
63. Rhodes B., Nash E.F., Tullis E., et al.: Prevalence of dyslipidemia in adults with CF. J. Cyst. Fibros., 2010; 9: 24–28
64. Radike K., Molz K., Holl R.W., Peoeter B., Hebestreit H., Ballmann M.: Prognostic relevance of hypoglycemia following an oral glucose challenge for cystic fibrosisrelated diabetes. Diabetes Care, 2011; 34: e43
65. Lanng S., Thorsteinsson B., Nerup J., Koch C.: Influence of the development of diabetes mellitus on clinical status in patients with cystic fibrosis. Eur. J. Pediatr., 1992; 151: 684–687
66. Milla C.E., Warwick W.J., Moran A.: Trends in pulmonary function in cystic fibrosis patients correlate with the degree of glucose intolerance at baseline. Am. J. Respir. Crit. Care Med., 2001; 162: 891–895
67. Nousia-Arvanitakis S., Galli-Tsinopoulou A., Karamouzis M.: Insulin improves clinical status of patients with cystic fibrosis-related diabetes mellitus. Acta Paediatr., 2001; 90: 515–519
68. Rolon M.A., Benali K., Munck A., et al.: CFRD: clinical impact of prediabetes and effects of insulin therapy. Acta Paediatr., 2001; 90: 860–867
69. White H., Morton A.M., Peckham D.G., Conway S.P.: Dietary intakes in adult patients with cystic fibrosis-do they achieve guidelines? J. Cyst. Fibros., 2004; 3: 1–7
70. Cheung M.S., Bridges N.A., Prasad S.A., et al.: Growth in children with cystic fibrosis-related diabetes. Pediatr. Pulmonol., 2009; 44: 1223–1225
71. Ripa P., Robertson I., Cowley D., Harris M., Master I.B., Cotterill A.M.: The relationship between insulin secretion, the insulin-like growth factor axis and growth in children with cystic fibrosis. Clin. Endocrinol., 2002; 56: 383–389
72. Moran A., Pekow P., Grover P., et al.: Insulin therapy to improve BMI in cystic fibrosis related diabetes without fasting hyperglycemia: results of the cystic fibrosis related diabetes therapy trial. Diabetes Care, 2009; 32: 1783–1788
73. Schmid K., Fink K., Holl R.W., Hebestreit H., Ballmann M.: Predictors for future cystic fibrosisrelated diabetes by oral glucose tolerance test. J. Cyst. Fibros., 2014; 13: 80–85
74. Geocobbe L.E., Nguyen R.H., Aguilera M.N., et al.: Effect of maternal cystic fibrosis genotype on diabetes in pregnancy. Obstet. Gynecol., 2012; 120: 1394–1399
75. Hameed S., Morton J., Jaffe A., et al.: Early glucose abnormalities in cystic fibrosis are preceded by poor weight gain. Diabetes Care, 2010; 33: 221–226
76. Brodsky J., Dougherty S., Ramkrishna M., Rubenstein R.C., Kelly A.: Elevation of 1-hour plasma glucose during oral glucose tolerance testing is associated with worse pulmonary function in cystic fibrosis. Diabetes Care, 2011; 34: 292–295
77. Mozzillo E., Raia V., Raggorusso V., et al.: Glucose derangements in very young children with cystic fibrosis and pancreatic insufficiency. Diabetes Care, 2012; 35: e78
78. Ode K.L., Frohnert B., Laguna T., et al.: Oral glucose tolerance testing in children with cystic fibrosis. Pediatr. Diabetes, 2010; 11: 487–492
79. Dobson L., Sheldon C.D., Hattersley A.T.: Conventional measures underestimate glycaemia in CF patients. Diabet. Med., 2004; 21: 691–696
80. Schiaffini R., Brufani C., Russo B., et al.: Abnormal glucose tolerance in children with cystic fibrosis: the predictive role of continuous glucose monitoring system. Eur. J. Endocrinol., 2010; 162: 705–710
81. Kolouskova S., Zemkova D., Bartosova J., et al.: Lowdose insulin therapy in patients with cystic fibrosis and early-stage insulinopenia prevents deterioration of lung function: a 30 year prospective study. J. Pediatr. Endocrinol. Metab., 2011; 24: 499–454
82. Sunni M., Bellin M.D., Moran A.: Exogenous insulin requirements do not differ between youth and adults with cystic fibrosis related diabetes. Pediatr. Diabetes, 2013; 14: 295–298
83. Konrad K., Thon A., Fritsch M., et al.: Comparison of CFRD with T1D based on aGerman/ Austrian pediatric diabetes registry. Diabetes Care, 2012; 35: 1–8
84. Onady G.M., Stolfi A.: Insulin and oral agents for managing cystic fibrosis-related diabetes. Cochrane Database Syst. Rev., 2013; 7: CD004 730 doi:10.1002/14 651 858.CD004 730.pub3
85. Nezer N., Shoseyov D., Kerem E., Zangen D.H.: Patients with cystic fibrosis and normoglycemia exhibit diabetic glucose tolerance during pulmonary exacerbation. J. Cyst. Fibros., 2010; 9: 199–204
86. Rasouli N., Seggelke S., Gibbs J., et al.: Cystic fibrosisrelated diabetes in adults: impatient management of 121 patients druing 410 admissions. J. Diabetes Sci. Technol., 2012; 6: 1038–1044
87. Mozzillo E., Franzese A., Valerio G., et al.: One year glargine treatment can improve the course of lung disease in children and adolescents with cystic fibrosis and early glucose derangements. Pediatr. Diabetes, 2009; 10: 162–167
88. Bizzarri C., Lucidi V., Ciampalini P., Bella S., Russo B., Cappa M.: Clinical effects of early treatment with insulin glargine in patients with cystic fibrosis and impaired glucose tolerance. J. Endocrinol. Invest., 2006; 29: RC1–RC4
89. Hameed S., Morton J.R., Field P.I., et al.: Once daily insulin detemir in cystic fibrosis with insulin deficiency. ADC, 2012; 97: 464–467
90. HAPO Study Cooperative Research Group: Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med., 2008; 358: 1991–2002